Стадия 1
Парафовеолярные изменения РПЭ : появляются точечные или пятнистые изменения пигментации ретинального пигментного эпителия (РПЭ).
Острота зрения и субъективные симптомы : чаще всего остаются нормальными.

Центральная ареолярная хориоидальная дистрофия (Central Areolar Choroidal Dystrophy; CACD) — это наследственная макулярная дистрофия, характеризующаяся четко очерченной атрофией хориоидеи и сетчатки в области макулы. Частота встречаемости низкая (около 1 на 100 000 человек), и она относится к редким заболеваниям.
Заболевание чаще всего начинается в возрасте 20–50 лет. Тип наследования преимущественно аутосомно-доминантный (АД), но также описаны аутосомно-рецессивные (АР) и спорадические случаи.
Наиболее частым причинным геном является PRPH2 (хромосома 17p13), кодирующий белок периферин-2. Периферин-2 необходим для формирования и стабильности дисков наружных сегментов фоторецепторов. Гаплонедостаточность (потеря функции одного аллеля) считается основным механизмом развития заболевания.
CACD имеет фенотипическое сходство с заболеваниями, вызванными мутациями гена ABCA4 (например, болезнь Штаргардта)1), и генетическое тестирование важно для дифференциальной диагностики. Системные осложнения не описаны.
ВМД обычно возникает после 60 лет и часто сопровождается друзами или экссудативными изменениями. CACD чаще возникает в молодом и среднем возрасте, характеризуется четко очерченными атрофическими очагами и не сопровождается друзами. Подтверждение мутации PRPH2 при генетическом тестировании позволяет диагностировать CACD.
Основным симптомом является двусторонняя центральная скотома (затруднение зрения в центре). Снижение остроты зрения может возникать рано, но иногда относительно хорошее зрение сохраняется до поздних стадий.
На обоих глазах наблюдаются симметричные макулярные поражения, которые классифицируются на 4 стадии. Зрительный нерв, сосуды сетчатки и периферическая сетчатка сохранены. Размер атрофической зоны обычно составляет 2–4 диаметра диска зрительного нерва (DD).
Стадия 1
Парафовеолярные изменения РПЭ : появляются точечные или пятнистые изменения пигментации ретинального пигментного эпителия (РПЭ).
Острота зрения и субъективные симптомы : чаще всего остаются нормальными.
Стадия 2
Гипопигментированная атрофия с нечеткими границами : за пределами фовеа появляется бледная зона атрофии.
Расширение изменений РПЭ : наблюдается атрофия с еще нечеткими границами.
Стадия 3
Атрофия РПЭ с четкими границами : за пределами фовеа формируется хорошо очерченная зона атрофии.
Сохранение фовеа : на этой стадии фовеа остается интактной, и острота зрения относительно сохранена.
Стадия 4
Полная атрофия, включающая фовеа: область атрофии распространяется на всю макулу, включая фовеа.
Тяжелое нарушение зрения: обширная потеря хориокапилляров, фоторецепторов и пигментного эпителия сетчатки приводит к значительному снижению остроты зрения.
Скорость прогрессирования сильно варьирует у разных людей. Обычно течение медленное, и в некоторых случаях до достижения 4-й стадии могут пройти десятилетия. Однако скорость прогрессирования может различаться в зависимости от генотипа, поэтому регулярное офтальмологическое обследование необходимо. Мультифокальная электроретинография, подробно описанная в разделе «Диагностика и методы обследования», является показателем раннего прогрессирования.
Основной причиной CACD является мутация в гене PRPH2. PRPH2 кодирует периферин-2, который участвует в формировании и стабилизации дисков наружных сегментов фоторецепторов. Мутации препятствуют поддержанию нормальной структуры наружных сегментов фоторецепторов.
Поскольку наследование аутосомно-доминантное (АД), вероятность передачи мутантного гена от родителя ребенку составляет 50%. При наличии семейного анамнеза заболевания рекомендуется обратиться за медико-генетическим консультированием.
На данный момент приобретенные факторы риска или факторы образа жизни, способствующие возникновению или прогрессированию заболевания, четко не идентифицированы.
CACD наследуется преимущественно аутосомно-доминантно, и генетический риск для детей пациента составляет 50%. Выявление мутации с помощью генетического теста позволяет поставить окончательный диагноз и провести семейный скрининг. Кроме того, поскольку в будущем возможна генная терапия, важно документировать мутацию.
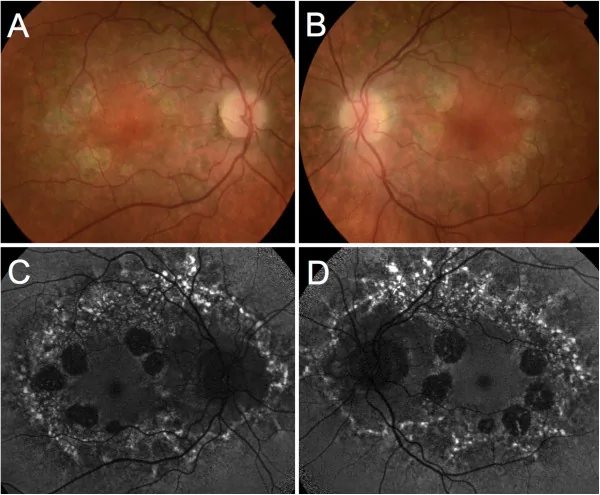
С помощью щелевой лампы и офтальмоскопа подтверждается наличие четко очерченной зоны атрофии в макуле. Оценивается обнажение хориоидальных сосудов и наличие пигментных отложений.
Результаты каждого исследования различаются в зависимости от стадии заболевания. В таблице ниже представлены характерные признаки по стадиям.
| Исследование | Ранние признаки | Признаки прогрессирующей стадии |
|---|---|---|
| FAF (аутофлуоресценция глазного дна) | Гиперфлуоресценция | Исчезновение флуоресценции (зона атрофии) |
| FA (флуоресцентная ангиография) | Парафовеолярная гиперфлуоресценция | Просвечивающее заполнение зоны атрофии |
| OCT | Утолщение POS-RPE | Исчезновение наружных слоев (фоторецепторов, RPE) |
Важно дифференцировать заболевания, проявляющиеся сходной макулярной атрофией.
| Дифференцируемое заболевание | Ключевые моменты дифференциации |
|---|---|
| Атрофическая ВМД | Друзы, нерегулярные границы, позднее начало |
| Болезнь Штаргардта | Темная хориоидея, мутация ABCA4 |
| Колбочковая дистрофия | Резкое снижение колбочкового ответа на ЭРГ, светобоязнь как основной симптом |
В настоящее время не существует установленного лечения CACD. Лечение направлено на контроль симптомов и поддержание качества жизни.
Реабилитация слабовидящих
Увеличительные стекла и светозащитные очки : Назначение вспомогательных средств для максимального использования остаточного зрения.
Профессиональное и бытовое обучение : Помощь в освоении навыков повседневной жизни с учетом нарушений зрения.
Генетическое консультирование
Информирование семьи : Объяснение аутосомно-доминантного наследования (50% риск передачи).
Генетическое тестирование : Выявление мутаций важно для окончательного диагноза и будущих вариантов лечения.
Генная терапия (исследовательская стадия)
Будущий кандидат на лечение : Исследования генной заместительной терапии, нацеленной на мутации PRPH2, продвигаются.
Текущее состояние : На данный момент она еще не доступна в качестве стандартного медицинского лечения.
Исследования генной терапии наследственных заболеваний сетчатки, включая те, которые связаны с PRPH2, ускоряются во всем мире. В Великобритании в 2014 году было проведено испытание генной терапии хориоидеремии, и изучается ее применение при заболеваниях, вызванных мутациями PRPH2. Однако в настоящее время она еще не установлена как стандартное лечение и находится на стадии исследований.
В зонах атрофии исчезают хориокапилляры, пигментный эпителий сетчатки (ПЭС) и фоторецепторы (палочки и колбочки). Количество клеток в наружном зернистом слое (НЗС) значительно уменьшается, и наружная пограничная мембрана (НПМ) вступает в прямой контакт с мембраной Бруха. Средние и крупные сосуды хориоидеи сохраняются в течение длительного времени даже на поздних стадиях заболевания.
Периферин-2 локализуется на краю (область ободка) дисков наружных сегментов фоторецепторов и играет важную роль в формировании, стабилизации и поддержании кривизны края дисков.
Гаплонедостаточность PRPH2 (потеря одной функциональной копии) нарушает нормальное формирование морфологии наружного сегмента. Структурный распад наружного сегмента приводит к апоптозу фоторецепторов, вызывая вторичную атрофию ПЭС и хориокапилляров.
Предполагается, что PRPH2 выполняет разные функции в палочках и колбочках. На модели мышей с нокаутом PRPH2 было сообщено, что синие колбочки дегенерируют медленнее, чем другие колбочки. Считается, что это различие между палочками и колбочками способствует разнообразию клинических фенотипов заболевания.
Мультифокальная электроретинография (mfERG) выявляет снижение функции даже в парафовеолярной области за пределами зоны атрофии, видимой при осмотре глазного дна. Это указывает на то, что поражения CACD распространяются шире, чем клинически видимая атрофия.
Исследования генной заместительной терапии и редактирования генов при наследственных заболеваниях сетчатки, включая мутации PRPH2, активно ведутся во всем мире. При родственном заболевании (хороидеремия) в 2014 году в Великобритании было проведено первое клиническое испытание генной терапии.
Анализ патогенности каждой мутации становится все более точным с развитием геномной медицины. Распространение полного секвенирования экзома (WES) значительно повысило точность диагностики 1), позволяя выявлять мутации у большего числа пациентов.
На молекулярном уровне все больше понимается, что PRPH2 выполняет разные функциональные роли в палочках и колбочках. Это знание важно для объяснения фенотипического разнообразия (дегенерация с преобладанием палочек или колбочек) и может повлиять на выбор будущих терапевтических мишеней.