Stade 1
Altérations de l’EPR parafovéolaire : des modifications pigmentaires ponctuelles ou en plaques de l’épithélium pigmentaire rétinien (EPR) apparaissent.
Acuité visuelle et symptômes subjectifs : souvent normaux et stables.

La dystrophie choriorétinienne aréolaire centrale (Central Areolar Choroidal Dystrophy ; CACD) est une dystrophie maculaire héréditaire caractérisée par une atrophie choroïdienne et rétinienne bien délimitée de la macula. Sa fréquence est faible (environ 1 personne sur 100 000) et elle est classée parmi les maladies rares.
L’apparition survient le plus souvent entre 20 et 50 ans. Le mode de transmission est principalement autosomique dominant (AD), mais des cas autosomiques récessifs (AR) et sporadiques ont également été rapportés.
Le gène le plus fréquemment impliqué est PRPH2 (chromosome 17p13), qui code la protéine périphérine-2. La périphérine-2 est essentielle à la formation et à la stabilité des disques des segments externes des photorécepteurs. L’haploinsuffisance (perte de fonction d’un allèle) est considérée comme le principal mécanisme pathogène.
La CACD présente des similitudes phénotypiques avec des maladies causées par des mutations du gène ABCA4 (comme la maladie de Stargardt)1), et les tests génétiques sont importants pour le diagnostic différentiel. Aucune complication systémique n’a été rapportée.
La DMLA survient généralement après 60 ans et s’accompagne souvent de drusen ou de modifications exsudatives. La CACD survient plus souvent chez les jeunes et les adultes d’âge moyen, se caractérise par des lésions atrophiques bien délimitées et ne présente pas de drusen. La confirmation d’une mutation PRPH2 par test génétique permet de diagnostiquer la CACD.
Le principal symptôme est un scotome central bilatéral (difficulté à voir au centre). Une baisse d’acuité visuelle peut être présente précocement, mais une bonne acuité visuelle peut parfois être maintenue jusqu’à un stade avancé.
Les deux yeux présentent des lésions maculaires symétriques, classées en 4 stades. Le nerf optique, les vaisseaux rétiniens et la rétine périphérique sont préservés. La taille de la zone atrophique est généralement de 2 à 4 diamètres papillaires (DD).
Stade 1
Altérations de l’EPR parafovéolaire : des modifications pigmentaires ponctuelles ou en plaques de l’épithélium pigmentaire rétinien (EPR) apparaissent.
Acuité visuelle et symptômes subjectifs : souvent normaux et stables.
Stade 2
Atrophie hypopigmentée aux limites floues : une zone d’atrophie pâle apparaît en dehors de la fovéa.
Extension des altérations de l’EPR : une atrophie aux limites encore imprécises est observée.
Stade 3
Atrophie de l’EPR aux limites nettes : une zone d’atrophie bien délimitée se forme en dehors de la fovéa.
Préservation fovéolaire : à ce stade, la fovéa est encore intacte et l’acuité visuelle relativement conservée.
Stade 4
Atrophie complète incluant la fovéa : la zone d’atrophie s’étend à toute la macula, y compris la fovéa.
Déficience visuelle sévère : la perte étendue de la choriocapillaire, des photorécepteurs et de l’EPR entraîne une diminution marquée de l’acuité visuelle.
La vitesse de progression varie considérablement d’un individu à l’autre. En général, l’évolution est lente, certains cas atteignant le stade 4 après plusieurs décennies. Cependant, la vitesse de progression peut différer selon le génotype, et une évaluation ophtalmologique régulière est indispensable. L’ERGmf, décrit en détail dans la section « Diagnostic et méthodes d’examen », est un indicateur de progression précoce.
La cause principale de la CACD est une mutation du gène PRPH2. PRPH2 code pour la périphérine-2, qui intervient dans la formation et la stabilisation des disques des segments externes des photorécepteurs. Les mutations empêchent le maintien de la structure normale des segments externes des photorécepteurs.
Étant donné qu’il s’agit d’une transmission autosomique dominante (AD), la probabilité de transmission du gène muté d’un parent à son enfant est de 50 %. En cas d’antécédents familiaux de la maladie, il est recommandé de consulter un conseiller en génétique.
Aucun facteur de risque acquis ou lié au mode de vie n’a été clairement identifié à ce jour comme favorisant l’apparition ou la progression de la maladie.
La CACD étant principalement autosomique dominante, le risque de transmission aux enfants est de 50 %. L’identification de la mutation par test génétique permet un diagnostic définitif et un dépistage familial. De plus, comme un traitement génétique futur pourrait être envisagé, il est important de documenter la mutation.
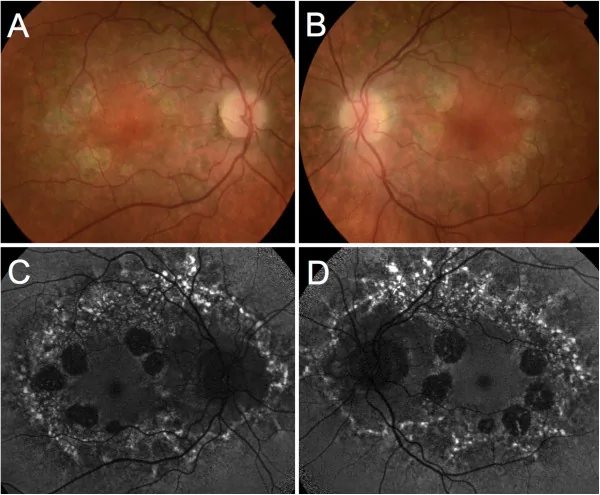
À l’aide d’une lampe à fente et d’un ophtalmoscope, on observe une zone d’atrophie bien délimitée au niveau de la macula. On évalue la présence de vaisseaux choroïdiens visibles et de dépôts pigmentaires.
Les résultats de chaque examen varient selon le stade de la maladie. Le tableau ci-dessous présente les caractéristiques typiques par stade.
| Examen | Signes précoces | Signes avancés |
|---|---|---|
| FAF (autofluorescence du fond d’œil) | Hyperfluorescence | Disparition de la fluorescence (zone atrophique) |
| FA (angiographie à la fluorescéine) | Hyperfluorescence juxtafovéolaire | Remplissage par transparence de la zone atrophique |
| OCT | Épaississement POS-RPE | Disparition des couches externes (photorécepteurs, RPE) |
Il est important de différencier les maladies présentant une atrophie maculaire similaire.
| Maladie différentielle | Points clés du diagnostic différentiel |
|---|---|
| DMLA atrophique | Drusen, limites irrégulières, apparition tardive |
| Maladie de Stargardt | Choroïde sombre, mutation ABCA4 |
| Dystrophie des cônes | Réduction marquée de la réponse des cônes à l’ERG, photophobie comme symptôme principal |
À l’heure actuelle, il n’existe pas de traitement établi pour la CACD. Le traitement vise à gérer les symptômes et à maintenir la qualité de vie.
Réadaptation en basse vision
Loupes et verres filtrants : Prescription d’aides pour maximiser l’utilisation de la fonction visuelle restante.
Formation professionnelle et à la vie quotidienne : Aide à l’acquisition de compétences pour la vie quotidienne adaptées à la déficience visuelle.
Conseil génétique
Information aux familles : Explication de la transmission autosomique dominante (risque de transmission de 50 %).
Tests génétiques : L’identification des mutations est importante pour le diagnostic définitif et les futures options thérapeutiques.
Thérapie génique (phase de recherche)
Futur candidat thérapeutique : La recherche sur la thérapie de remplacement génique ciblant les mutations PRPH2 progresse.
Situation actuelle : À ce stade, elle n’est pas encore disponible en tant que traitement médical standard.
La recherche sur la thérapie génique pour les maladies rétiniennes héréditaires, y compris celles impliquant PRPH2, s’accélère dans le monde entier. Au Royaume-Uni, un essai de thérapie génique pour la choroidérémie a été réalisé en 2014, et l’application aux maladies liées à des mutations de PRPH2 est également étudiée. Cependant, à l’heure actuelle, elle n’est pas encore établie comme traitement standard et en est au stade de la recherche.
Dans les zones atrophiques, la choriocapillaire, l’EPR et les photorécepteurs (bâtonnets et cônes) disparaissent. Le nombre de cellules dans la couche nucléaire externe (ONL) diminue considérablement, et la membrane limitante externe (OLM) entre en contact direct avec la membrane de Bruch. Les vaisseaux choroïdiens de taille moyenne à grande sont préservés pendant longtemps même à des stades avancés de la maladie.
La périphérine-2 est localisée au bord (région du rim) des disques des segments externes des photorécepteurs et joue un rôle essentiel dans la formation, la stabilisation et le maintien de la courbure des disques.
L’haploinsuffisance de PRPH2 (perte d’une copie fonctionnelle) perturbe la formation normale de la morphologie du segment externe. La rupture structurelle du segment externe conduit à l’apoptose des photorécepteurs, entraînant une atrophie secondaire de l’EPR et de la choriocapillaire.
Il est suggéré que PRPH2 a des fonctions différentes dans les bâtonnets et les cônes. Dans un modèle de souris knockout pour PRPH2, il a été rapporté que les cônes bleus dégénèrent plus lentement que les autres cônes. On pense que cette différence entre bâtonnets et cônes contribue à la diversité des phénotypes cliniques de la maladie.
L’électrorétinographie multifocale (mfERG) détecte une diminution de la fonction même dans la région parafovéale au-delà de la zone atrophique visible à l’examen du fond d’œil. Cela indique que les lésions de la CACD s’étendent plus largement que l’atrophie cliniquement visible.
Des recherches sur la thérapie génique de remplacement et l’édition génique pour les maladies rétiniennes héréditaires, y compris les mutations PRPH2, sont menées dans le monde entier. Pour une maladie apparentée (choroidérémie), le premier essai clinique de thérapie génique a été réalisé au Royaume-Uni en 2014.
L’analyse de la pathogénicité de chaque mutation est de plus en plus précise avec les progrès de la médecine génomique. La généralisation du séquençage de l’exome entier (WES) a considérablement amélioré la précision du diagnostic 1), permettant d’identifier les mutations chez un plus grand nombre de patients.
Il est de plus en plus compris au niveau moléculaire que PRPH2 a des rôles fonctionnels différents dans les bâtonnets et les cônes. Cette connaissance est importante pour expliquer la diversité des phénotypes (dégénérescence à prédominance de bâtonnets ou de cônes) et pourrait influencer le choix des cibles thérapeutiques futures.