Predisposition and epidemiology
Age and sex: Mean age 30.6 years, 62.3% female, mostly unilateral
Bilateral involvement: Rare at 9.8%
Recurrence rate: Rare at 3%

Acute retinal pigment epitheliitis (ARPE) is a rare idiopathic, self-limiting inflammatory retinal disease first reported by Alex E. Krill and August F. Deutman in 1972 1, 2, 4). It is also called Krill disease after the discoverer. It is classified as one of the white dot syndromes.
It predominantly affects healthy young adults, with a mean age of 30.6 ± 10.7 years (range 16–55 years) and a female predominance (62.3%) 2). It is mostly unilateral, with bilateral involvement in only 9.8% of cases. Recurrence is rare (3%) 2).
An association with viral infection has been reported in 25.9% of cases 2). Cold-like symptoms may precede the onset. Drug associations include D2 dopamine receptor agonists (bromocriptine, cabergoline) and intravenous bisphosphonates 1). Cases after COVID-19 and influenza vaccination have also been reported 4).
Whether ARPE is an independent disease or a subtype of other diseases such as MEWDS, pachychoroid, or AMN remains a topic of ongoing debate 1).
It is an extremely rare disease. A literature review identified only 61 cases from 29 papers, and the exact incidence is unknown. Each time a rare case is reported, the disease concept is re-evaluated.
Onset is acute, presenting with the following symptoms.
Predisposition and epidemiology
Age and sex: Mean age 30.6 years, 62.3% female, mostly unilateral
Bilateral involvement: Rare at 9.8%
Recurrence rate: Rare at 3%
Main subjective symptoms
Blurred vision and metamorphopsia: Characterized by acute onset
Central scotoma: Corresponds to macular lesions
Color vision abnormalities: Blue → green, yellow → beige, red → gray
Visual prognosis
Initial visual acuity: ≈20/40 (corrected visual acuity)
Final visual acuity: ≈20/20
Complete recovery rate: Approximately 89% achieve 20/20 within 2 months
No anterior segment abnormalities or intraocular inflammation are observed4). Fundus examination reveals fine pigment spots surrounded by a yellowish-white hypopigmented halo in the macula2, 4). The lesion has a characteristic appearance with a dark central core and a grayish-white halo around it.
Initial visual acuity is approximately 20/40, and in most cases, final visual acuity recovers to about 20/202). Electrophysiological tests show reduced P50 wave amplitude in pERG and decreased response density in mfERG, reflecting macular dysfunction1).
Approximately 89% recover to 20/20 within 2 months. However, cases with persistent EZ (ellipsoid zone) disruption on OCT for more than 12 months or extensive lesions reaching the ONL (outer nuclear layer) have been reported to have incomplete recovery (see OCT findings).
The cause of ARPE is unknown, and most cases are considered idiopathic2). Currently known risk factors are as follows.
Viral infection (most common) An association with viral infection has been reported in 25.9% of cases2). Cold-like symptoms may precede the onset.
After vaccination Onset 31 days after the second dose of COVID-19 vaccine has been reported. In that case, the patient had low-grade fever (37.3–37.5°C) and joint pain for 2 days after vaccination, and had also received an influenza vaccine 5 days earlier 4). Bolletta et al. reported cases of ocular inflammation occurring 28–30 days after COVID-19 vaccination 4).
Drug-induced Onset during treatment for hyperprolactinemia with D2 dopamine receptor agonists (bromocriptine, cabergoline) has been reported 1). Cases after intravenous bisphosphonate administration also exist 1).
Onset after COVID-19 or influenza vaccination has been reported 4). The immune response triggered by vaccination may be a trigger, but a causal relationship has not yet been established.
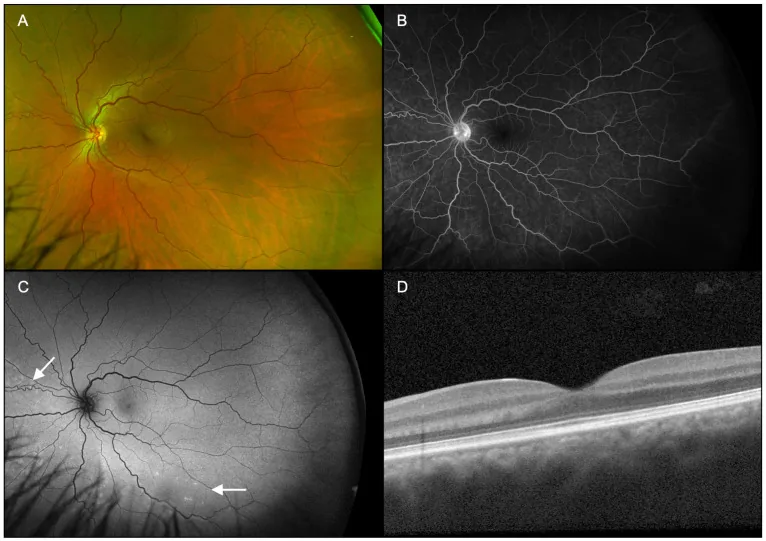
Diagnosis of ARPE is made by combining multiple imaging examinations. Characteristic findings of each examination are shown below.
OCT (optical coherence tomography) is the most important diagnostic and follow-up tool.
The frequency of involvement and healing duration for each retinal layer are shown below.
| Affected layer | Frequency | Healing duration |
|---|---|---|
| IZ (inner/outer segment junction) | 100% | 9.1±8.3 weeks |
| EZ (ellipsoid zone) | 95.6% | 7.2±5.2 weeks |
| ELM (external limiting membrane) | 35.6% | 5.1±4.8 weeks |
The main findings are as follows.
Characteristic findings of each imaging modality are shown below.
| Test | Characteristic Findings | Positive Rate |
|---|---|---|
| FA | Transmitted hyperfluorescence, no leakage | 83.6% |
| ICGA | Cockade-shaped hyperfluorescent halo | 84.6% |
| Fundus autofluorescence | Mild hyperautofluorescence in the lesion | 40% |
It is important to differentiate from the following diseases 3, 4).
ARPE is a self-limiting disease, with spontaneous resolution expected within 6 to 12 weeks. Basically, active treatment is not necessary 2).
Observation is the mainstay Many reports show natural recovery of vision without treatment. In the case by Kilic (2021), vision recovered to 20/20 after one month without treatment 1). In the post-vaccine ARPE case by Sasajima et al. (2022), vision recovered to 1.5 (equivalent to 20/13) after 5 weeks without treatment 4).
Efficacy of steroids is not established There are reports of cases recovering after 6 months with oral steroids 3), but also reports that the steroid-treated group had slower visual recovery than the untreated group. The basic policy is to decide on treatment initiation and follow-up observation.
Basically, treatment is not necessary, and spontaneous resolution occurs within 6 to 12 weeks. There is no evidence that steroid administration accelerates recovery; rather, there are reports that it may delay recovery. Regular follow-up with OCT is recommended.
The pathophysiology of ARPE is not fully understood, but several hypotheses have been proposed.
MerTK Deficiency Hypothesis
POS phagocytosis disorder: MerTK deficiency → POS accumulation → outer layer hyperreflectivity → photoreceptor degeneration
Three-step disorder: recognition/binding (αvβ5 + MFG-E8), uptake (MerTK activation), lysosomal digestion
Related disease: MerTK mutations cause retinitis pigmentosa in humans
Choroidal ischemia hypothesis
Circadian rhythm disruption hypothesis
Dopamine system disturbance: D2 receptor agonists suggested to cause ARPE
POS shedding abnormality: Retinal circadian clock controls POS disc shedding
D2 receptor stimulation: Inhibits cone-rod gap junctions → reduced photoreceptor light sensitivity
Phagocytosis of photoreceptor outer segments (POS) by RPE proceeds in three steps 2).
Acute transient MerTK deficiency is presumed to be central to the pathogenesis of ARPE 2). MerTK mutations are known to cause retinitis pigmentosa in humans, suggesting a disease continuum with ARPE.
Flow voids in the choriocapillaris on OCTA suggest a mechanism similar to APMPPE 3). A mechanism has been proposed in which primary choroidal ischemia secondarily damages photoreceptors and the Henle fiber layer (HFL), and OCT findings indicate that the main lesion of ARPE is in the outer retinal layers rather than in the “pigment epitheliopathy.”
Although the disease is named “pigment epitheliopathy,” OCT findings show that the main location is the outer retinal layers such as the IZ (inner/outer segment junction) and EZ (ellipsoid zone). The IZ is affected in 100% and the EZ in 95.6% of cases, while abnormalities of the RPE/Bruch membrane are limited to 8.9% 2). RPE changes are likely secondary.
Whether ARPE is an independent disease entity remains under debate. There are opposing positions: one denies its independent disease nature as a “diagnostic myth,” and the other supports it as “not a diagnostic myth” 1, 3). Its relationship with MEWDS, AMN, and APMPPE, as well as the possibility of inclusion in the pachychoroid spectrum, are being considered.
Sen et al. (2025) reported the ASHH (Angular Sign of Henle Fiber Layer Hyperreflectivity) in presumed ARPE cases 3).
Sen et al. (2025) identified ASHH and choriocapillaris flow voids on OCTA in presumed ARPE cases, suggesting that choroidal ischemia may be involved in the pathogenesis of ARPE 3). It was proposed to position ARPE as one of the differential diagnoses of pachychoroid pigment epitheliopathy.
Sasajima et al. (2022) reported a case of ARPE that developed 31 days after the second dose of COVID-19 vaccine 4). The EZ/IZ disruption on OCT was repaired within 5 weeks, and visual acuity recovered to 1.5. It has been hypothesized that the post-vaccination immune response targets the RPE or outer retina, triggering inflammation.
The MerTK deficiency hypothesis remains at the hypothesis stage and requires direct verification in human cases 2). However, the fact that ARPE can also be induced by drugs (D2 receptor agonists) has drawn attention to the mechanism by which dopamine-mediated retinal circadian rhythm disruption disturbs POS shedding 1, 2).