Kobalamin C eksikliği (cblC tipi), hücre içi B12 vitamini (kobalamin) metabolizmasının doğuştan gelen bir metabolik hastalığıdır. Resmi adı metilmalonik asidüri ve homosistinüri cblC tipidir (OMIM #277400). Tüm cbl metabolizma bozukluklarının yaklaşık %80’ini oluşturan en sık görülen tiptir.
Kalıtım şekli otozomal resesiftir. Nedeni MMACHC genindeki (1p34.2) mutasyondur. MMACHC proteini, hücre içi kobalamin metabolizmasının erken aşamalarından sorumludur ve işlev kaybı, hem adenosilkobalamin (AdoCbl) hem de metilkobalamin (MeCbl) sentezinin bozulmasına yol açar.
Tahmini insidans 1:100.000 ila 1:200.000 olarak bildirilmiştir3, 4), ancak Çin’in Shandong eyaletinde yapılan bir çalışmada 1:3.920 gibi yüksek bir sıklık rapor edilmiştir. ABD’de 2000’li yılların başından itibaren yenidoğan taramasının (NBS) bir parçası haline gelmiştir4).
Başlangıç yaşına göre erken başlangıçlı (yaşamın ilk yılı, tüm vakaların %86-88’i) ve geç başlangıçlı (1 yaş sonrası) olarak ikiye ayrılır. Geç başlangıçlı 160’tan fazla vaka ve erişkin başlangıçlı (18 yaş üstü) 30’dan fazla vaka bildirilmiştir3).
QYenidoğan taramasında kobalamin C eksikliği tespit edilebilir mi?
A
Kanda yüksek C3 propiyonil karnitin düzeyi göstergesi ile NBS sayesinde erken teşhis mümkündür. Bununla birlikte, geç başlangıçlı ve hafif vakalar NBS’den kaçabilir4); bu nedenle klinik şüphe durumunda tarama normal olsa bile plazma Hcy ve idrar MMA ek testleri gereklidir.
Beyin MRG’sinde beyaz cevher lezyonları: Bazı geç başlangıçlı tiplerde görülür3).
Hipertansif retinopati: Böbrek komplikasyonu (trombotik mikroanjiyopati) olan ciddi vakalarda ortaya çıkabilir2).
Sistemik komplikasyonlar arasında metabolik asidoz, hipotoni, mikrosefali, kardiyomiyopati (%50)1) ve renal trombotik mikroanjiyopati (TMA)2) bilinmektedir.
QGeç başlangıçlı tipte oküler komplikasyonlar olur mu?
A
Geç başlangıçlı tipte, erken başlangıçlı tipteki gibi ciddi makülopati ve retina dejenerasyonu nadirdir ve çoğu vakada fundus normaldir3, 4). Ancak böbrek komplikasyonu olan ciddi vakalarda hipertansif retinopati görülebilir2). Nörolojik semptomlar genellikle tedaviye iyi yanıt verir, ancak kalıcı görme bozukluğu olan vakalar da bildirilmiştir3).
Kobalamin C eksikliği, MMACHC genindeki mutasyonlara bağlı otozomal resesif bir hastalıktır. 80’den fazla mutasyon bildirilmiştir ve mutasyon tipine göre fenotipler büyük farklılık gösterir.
Başlıca mutasyonların sıklığı ve karşılık gelen fenotipler aşağıda gösterilmiştir.
Mutasyon
Sıklık/soy
Ana fenotip
c.271dupA
%40-61 · Avrupa kökenli
Erken başlangıçlı · En şiddetli
c.394C>T
Yaklaşık %20
Geç başlangıçlı tip
c.609G>A
%48-55 · Doğu Asya kökenli
Bebeklikten çocukluğa
Diğer başlıca mutasyonlar şunlardır:
c.331C>T: Fransız-Kanada kökenlilerde %5-91).
c.566G>A: Geç başlangıçlı tip ile ilişkili3).
c.484G>T: Bebeklik döneminde çoklu organ tutulumu ile ilişkili2).
c.271dupA + c.449T>A (bileşik heterozigot): Geç başlangıçlı seyir gösteren olgular bildirilmiştir4).
Ayrıca, PRDX1 gen mutasyonuna bağlı MMACHC’nin epigenetik susturulmasının neden olduğu “epi-cblC” de mevcuttur ve sekonder cblC benzeri bir fenotipe yol açar.
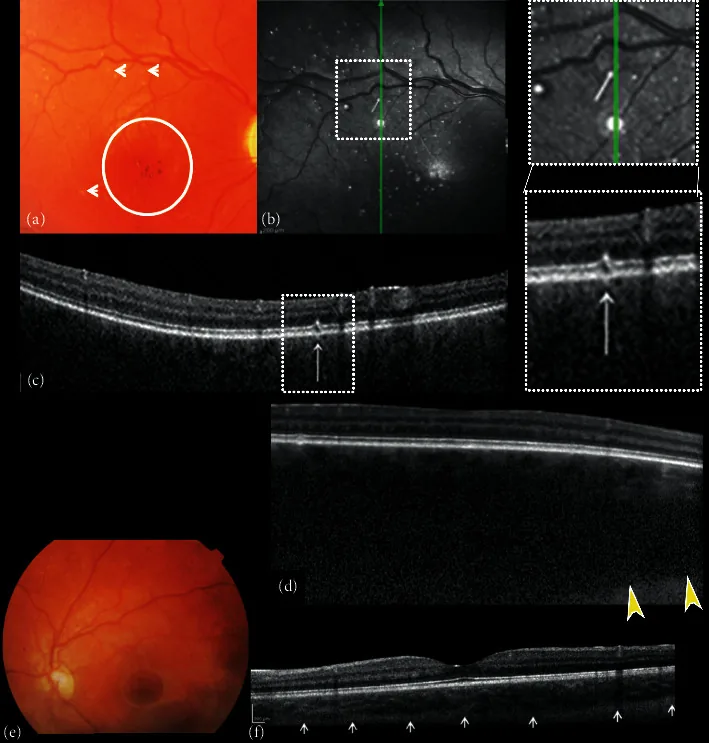
Sağ ve sol gözün fundus renkli fotoğrafları ve spektral-domain optik koherens tomografi (SDOCT) bileşimi. (a) Sağ fundus görüntüsü, optik diskte çukurlaşma ve solukluk, tessellasyon yokluğu, diffüz koroidal hemanjiyom, foveal refleksin olmadığı foveal alanda hipo-hiperpigmentasyon (daire) ve küçük beyaz nokta şeklinde “mikrodrüzen benzeri” değişiklikler (oklar) gösterir. (b) Sağ gözün yakın kızılötesi yansıması (NIR), oftalmoskopi ile gözlenen arka kutuptaki küçük beyaz nokta şeklindeki “mikrodrüzen benzeri” değişikliklere karşılık gelen, hiporeflektif bir halka ile çevrili çoklu hiperreflektif noktalar gösterir. Hiperreflektif noktalar üzerindeki B-taraması kesitsel SDOCT taraması (c), retina pigment epiteli (RPE)-fotoreseptör tabakasının fokal değişikliklerini gösterir.
Yenidoğan taraması (NBS): Tandem kütle spektrometrisi ile C3 propiyonil karnitin yüksekliği gösterge olarak kullanılır. Ancak hafif ve geç başlangıçlı vakalar gözden kaçabilir4).
Diyete bağlı B12 eksikliği, pernisiyöz anemi, diğer cbl metabolizma bozuklukları (cblD, cblF vb.) ve MTHFR eksikliğinden ayırt edilmesi önemlidir 3, 4). Yüksek Hcy ve yüksek MMA kombinasyonu cblC için karakteristiktir; yalnızca yüksek Hcy (MMA normal) MTHFR eksikliği vb. durumları düşündürür.
QSerum B12 normal iken cblC olasılığı var mıdır?
A
Evet. Geç başlangıçlı cblC’de serum B12 sıklıkla normal aralıktadır 3, 4) ve sadece B12 normal diye dışlanmamalıdır. Nörolojik veya psikiyatrik semptomları olan genç hastalarda mutlaka plazma Hcy ve MMA ölçülmeli, yüksekse genetik testle MMACHC analiz edilmelidir.
Parenteral uygulama esastır. Yaklaşık doz: 1 mg/gün veya 0.3 mg/kg/gün intramüsküler (IM) enjeksiyon 4). Bildirilen dozlar:
Ailliet ve ark. (2022) geç başlangıçlı bir erişkin vakada 25 mg/gün subkutan (SC) enjeksiyon uygulamış ve nörolojik semptomlarda düzelme gözlemlemiştir 3). Goyne ve ark. (2023) bir vakada 5 mg/gün IM ile başlamıştır 4). Hjalmarsson ve ark. (2024) ağır kalp yetmezliği olan bir vakada 30 mg/gün intravenöz (IV) uygulama yapmıştır 1).
Tedavi hedefi MMA ve Hcy düzeylerini düşürmek ve metabolik göstergeleri normalleştirmektir. Uygun tedavi ile Hcy’de belirgin düşüş sağlanır:
Akar ve ark. (2024) tarafından bildirilen böbrek TMA’sı olan ciddi bir vakada, tedavi başlangıcından sonra Hcy 1700,5’ten 4,6 µmol/L’ye normalize olmuştur 2). Hjalmarsson ve ark. (2024) tarafından bildirilen kardiyomiyopatili bir vakada, MMA 97’den 8,1 mmol/mol’e ve Hcy 91’den 31,8 µmol/L’ye iyileşmiştir 1).
Geç başlangıçlı tipte tedaviye yanıt nispeten iyidir 4), ancak görme fonksiyonundaki iyileşme sınırlı olabilir 3). Erken başlangıçlı tipte, uygun tedaviye rağmen birçok hasta ergenlik döneminde yasal körlüğe ulaşır.
Kalp: Beta blokerler vb. ile kalp yetmezliği tedavisi. Ciddi vakalarda LVAD (sol ventrikül destek cihazı) veya kalp nakli de düşünülür 1).
Böbrek: ACE inhibitörleri veya ARB ile böbrek koruması 2).
QTedavi göz semptomlarını iyileştirir mi?
A
Erken başlangıçlı tipte, metabolik göstergeler düzelse bile, makülopati ve retina dejenerasyonunun görme fonksiyonu prognozu sıklıkla kötüdür ve ergenlikte yasal körlüğe ulaşan vakalar bildirilmiştir. Geç başlangıçlı tipte nörolojik semptomlara tedavi yanıtı nispeten iyidir, ancak kalan görme bozukluğu sadece sınırlı olarak düzelir 3). Erken tanı ve erken tedavi başlangıcı en iyi prognozu sağlayabilir.
MMACHC proteini, lizozomda kobalamin-haptokorin (R) kompleksine bağlanır ve kobalaminden R grubunu ayırarak hücre içinde kullanılabilir ortak bir kobalamin ara ürünü oluşturur. Bu ara ürün daha sonra iki yola ayrılır.
Mitokondriyal yol: AdoCbl sentezler ve metilmalonil-CoA mutazın koenzimi olarak işlev görür.
Sitoplazmik yol: MeCbl sentezler ve metiyonin sentazın koenzimi olarak işlev görür.
MMACHC işlev kaybı nedeniyle her iki yol da bozulursa:
Göz dokusu üzerindeki etkiler açısından, metiyonin düşüklüğüne bağlı glutatyon (GSH) sentez bozukluğunun retina pigment epitelinde (RPE) oksidatif stres savunmasını azalttığı ve makula dejenerasyonuna katkıda bulunduğu düşünülmektedir. Yüksek Hcy’ye bağlı vasküler endotel hasarının retina ve koroid dolaşımını bozduğu mekanizma da öne sürülmüştür.
7. Güncel Araştırmalar ve Gelecek Perspektifler (Araştırma Aşamasındaki Raporlar)
Hjalmarssон ve ark. (2024), kardiyomiyopati ile komplike olan bir cblC hastasında LVAD (sol ventrikül destek cihazı) implantasyonu ve ardından kalp nakli yapılan dünyadaki ilk vakayı bildirdi1). Yüksek doz OH-Cbl (30 mg/gün IV), betain, folinik asit ve L-karnitin metabolik tedavisi ile birlikte, metabolik göstergelerde iyileşme (MMA 97→8.1, Hcy 91→31.8) ve iyi bir sonuç elde edildi.
Kalp naklinden sonra bile cblC için metabolik tedavinin devamının gerekli olduğu gösterildi1).
Yenidoğan Taramasının Sınırlamaları ve Geç Başlangıçlı Tipin Tanısının İyileştirilmesi
Geç başlangıçlı ve hafif vakaların yenidoğan taramasında (NBS) gözden kaçabileceği gösterilmiştir4) ve nörolojik ve psikiyatrik semptomları olan genç erişkinlerde B12 normal olsa bile Hcy ve MMA ölçümünün önemi vurgulanmıştır3, 4).
PRDX1 gen mutasyonuna bağlı MMACHC’nin epigenetik susturulmasıyla oluşan «epi-cblC» rapor edilmiştir. Gen mutasyonu olmasa bile cblC benzeri fenotip ortaya çıkabileceğinden, tanı algoritmasının yeniden gözden geçirilmesi gerekmektedir.
Ailliet ve ark. (2022), geç başlangıçlı cblC’de aile taraması yoluyla asemptomatik homozigotları erken teşhis etme deneyimlerini bildirmiş ve proband tanısı sonrası aktif aile taramasının önemini vurgulamıştır3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.