Дефицит кобаламина C (тип cblC) — это врожденное нарушение внутриклеточного метаболизма витамина B12 (кобаламина). Полное название — метилмалоновая ацидурия и гомоцистинурия, тип cblC (OMIM #277400). Это самый частый тип среди всех нарушений метаболизма кобаламина, составляющий около 80% случаев.
Тип наследования — аутосомно-рецессивный. Причина — мутации в гене MMACHC (1p34.2). Белок MMACHC участвует в ранних этапах внутриклеточного метаболизма кобаламина; потеря его функции приводит к нарушению синтеза как аденозилкобаламина (AdoCbl), так и метилкобаламина (MeCbl).
Предполагаемая частота встречаемости составляет от 1:100 000 до 1:200 000 3, 4), однако исследование в китайской провинции Шаньдун сообщило о высокой частоте 1:3 920. В США с начала 2000-х годов это заболевание включено в неонатальный скрининг (NBS) 4).
По возрасту начала выделяют раннюю форму (в течение первого года жизни, 86–88% всех случаев) и позднюю форму (после 1 года). Сообщается о более чем 160 случаях поздней формы и более чем 30 случаях с началом во взрослом возрасте (≥18 лет) 3).
QМожно ли выявить дефицит кобаламина C при неонатальном скрининге?
A
NBS на основе повышенного уровня C3-пропионилкарнитина в крови позволяет выявить заболевание на ранней стадии. Однако поздние или легкие формы могут быть пропущены при скрининге 4); при клиническом подозрении, несмотря на нормальный скрининг, необходимы дополнительные тесты (Hcy в плазме, MMA в моче).
Клиническая картина значительно различается в зависимости от возраста начала заболевания.
Глазные симптомы ранней формы:
Нистагм: Самый ранний глазной признак. Наблюдается примерно у 70–76% пациентов.
Снижение остроты зрения: Прогрессирует с младенчества и, даже при соответствующем лечении, часто приводит к юридической слепоте в подростковом возрасте.
Косоглазие: Часто сочетается с нистагмом.
Основные симптомы поздней формы (внеглазные):
Слабость и онемение ног: Вследствие поражения задних столбов спинного мозга4).
Нарушения походки и психические симптомы: Сообщается при случаях с началом во взрослом возрасте3, 4).
Данные офтальмоскопии значительно различаются между ранней и поздней формами.
Ранняя форма
Макулопатия: Самый характерный признак. Может появляться с 35-го дня жизни.
Макулопатия типа «бычий глаз»: Характерный рисунок с центральной хориоретинальной атрофией и пигментным кольцом.
Изменения, подобные пигментному ретиниту: Обширная дегенерация сетчатки.
Атрофия зрительного нерва: Наблюдается в запущенных случаях.
Флюоресцентная ангиография глазного дна (ФАГ) : снижение аутофлюоресценции в фовеа и повышение по краям.
Поздняя форма
Часто нормальное глазное дно : часто без тяжелых глазных проявлений3, 4).
МРТ спинного мозга : характерны гиперинтенсивные очаги в задних столбах4).
МРТ головного мозга: очаги в белом веществе : наблюдаются при некоторых поздних формах3).
Гипертоническая ретинопатия : может возникать при тяжелых случаях с почечными осложнениями (тромботическая микроангиопатия)2).
Системные осложнения включают метаболический ацидоз, мышечную гипотонию, микроцефалию, кардиомиопатию (50%)1) и почечную тромботическую микроангиопатию (ТМА)2).
QВозникают ли глазные осложнения при поздней форме?
A
При поздней форме тяжелая макулопатия и дегенерация сетчатки, как при ранней форме, редки, глазное дно часто нормальное3, 4). Однако при тяжелых случаях с почечными осложнениями может развиться гипертоническая ретинопатия2). Неврологические симптомы часто хорошо поддаются лечению, но описаны случаи с остаточными нарушениями зрения3).
Дефицит кобаламина C — аутосомно-рецессивное заболевание, вызванное мутациями в гене MMACHC. Описано более 80 мутаций, фенотип значительно варьирует в зависимости от типа мутации.
Частота основных мутаций и соответствующие фенотипы приведены ниже.
Мутация
Частота / Происхождение
Основной фенотип
c.271dupA
40–61% у европейцев
Раннее начало, наиболее тяжелая форма
c.394C>T
Около 20%
Поздняя форма
c.609G>A
48–55% у восточных азиатов
Младенческий–детский возраст
Другие основные мутации:
c.331C>T: 5–9% у франкоканадцев1).
c.566G>A: связана с поздней формой3).
c.484G>T: связана с полиорганной недостаточностью у младенцев2).
c.271dupA + c.449T>A (компаунд-гетерозигота): описаны случаи с поздней формой4).
Также существует «epi-cblC», вызванный эпигенетическим сайленсингом MMACHC из-за мутации гена PRDX1, приводящий к вторичному cblC-подобному состоянию.
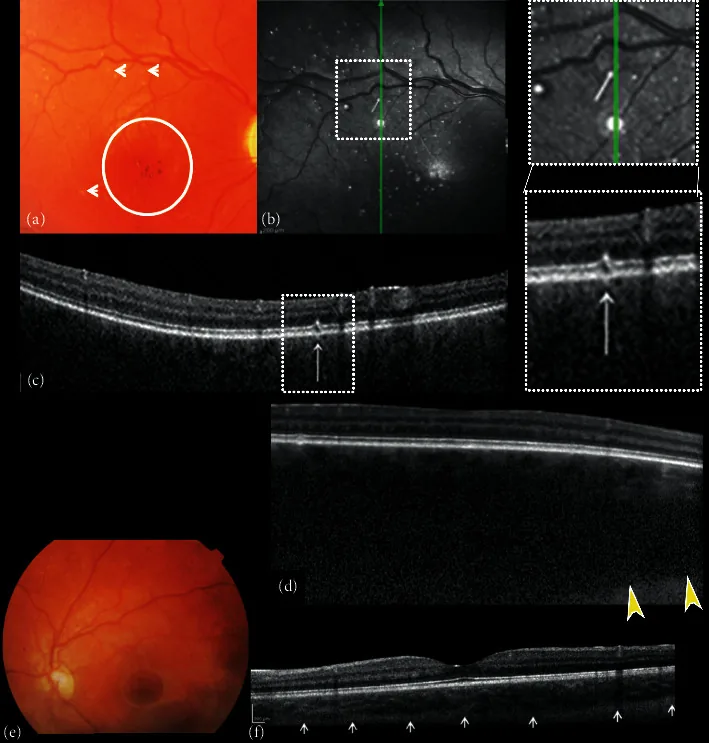
Композит цветных фотографий глазного дна и спектральной оптической когерентной томографии (SDOCT) правого и левого глаза. (a) Изображение правого глазного дна показывает экскавацию и бледность диска зрительного нерва, отсутствие тесселяции, диффузную хориоидальную гемангиому, гипо-гиперпигментацию фовеальной области с отсутствием фовеального рефлекса (круг) и мелкие белые точечные «микродрузоподобные» изменения (стрелки). (b) Инфракрасное отражение (NIR) правого глаза показывает множественные гиперрефлективные точки, окруженные гипорефлективным кольцом, соответствующие мелким белым точечным «микродрузоподобным» изменениям заднего полюса, наблюдаемым при офтальмоскопии. B-скановое поперечное сечение SDOCT (c) на гиперрефлективных точках показывает очаговые изменения слоя фоторецепторов пигментного эпителия сетчатки (RPE).
Неонатальный скрининг (NBS) : Повышенный уровень C3-пропионилкарнитина методом тандемной масс-спектрометрии является показателем. Однако легкие и поздно начинающиеся формы могут быть пропущены 4).
Важно дифференцировать алиментарный дефицит B12, пернициозную анемию, другие нарушения метаболизма кобаламина (cblD, cblF и др.) и дефицит MTHFR3, 4). Сочетание повышенного Hcy и повышенного MMA характерно для cblC, тогда как только повышенный Hcy (нормальный MMA) указывает на дефицит MTHFR и др.
QВозможен ли cblC при нормальном уровне витамина B12 в сыворотке?
A
Да. При cblC с поздним началом сывороточный B12 часто находится в пределах нормы3, 4); нормальный B12 сам по себе не исключает диагноз. У молодых пациентов с неврологическими или психиатрическими симптомами необходимо измерить Hcy и MMA в плазме, и если они повышены, провести генетический анализ гена MMACHC.
Парентеральное введение является стандартом. Ориентировочная доза: 1 мг/сут или 0,3 мг/кг/сут внутримышечно (ВМ)4). Сообщаемые дозы:
Ailliet и соавт. (2022) вводили 25 мг/сут подкожно (ПК) взрослому пациенту с поздним началом, что привело к улучшению неврологических симптомов3). Goyne и соавт. (2023) начинали с 5 мг/сут ВМ4). Hjalmarsson и соавт. (2024) вводили 30 мг/сут внутривенно (ВВ) пациенту с тяжелой сердечной недостаточностью1).
Цели лечения — снижение MMA и Hcy и нормализация метаболических показателей. Адекватное лечение приводит к значительному снижению Hcy:
Акар и др. (2024) сообщают о тяжелом случае с почечной ТМА, где Hcy после начала лечения нормализовался с 1700,5 до 4,6 мкмоль/л2). Хьялмарссон и др. (2024) описывают случай с кардиомиопатией, где MMA улучшился с 97 до 8,1 ммоль/моль, а Hcy — с 91 до 31,8 мкмоль/л1).
При поздних формах ответ на лечение относительно хороший4), но восстановление зрительной функции может быть ограниченным3). При ранних формах, несмотря на адекватное лечение, многие пациенты достигают юридической слепоты в подростковом возрасте.
Сердце: Лечение сердечной недостаточности бета-блокаторами и др. В тяжелых случаях может рассматриваться LVAD (устройство вспомогательного кровообращения левого желудочка) или трансплантация сердца1).
Почки: Защита почек с помощью ингибиторов АПФ или БРА2).
QУлучшаются ли глазные симптомы при лечении?
A
При ранних формах, даже при улучшении метаболических показателей, прогноз зрения при макулопатии и дегенерации сетчатки часто плохой, и сообщается о случаях юридической слепоты в подростковом возрасте. При поздних формах ответ неврологических симптомов на лечение относительно хороший, но остаточные нарушения зрения восстанавливаются лишь ограниченно3). Ранняя диагностика и раннее начало лечения могут обеспечить наилучший прогноз.
Белок MMACHC связывается в лизосоме с комплексом кобаламин-гаптокорин (R), отщепляет R-группу от кобаламина и генерирует общий промежуточный продукт кобаламина, доступный для использования в клетке. Затем этот промежуточный продукт расходится по двум путям.
Митохондриальный путь: Синтез AdoCbl, который действует как кофермент метилмалонил-КоА-мутазы.
Цитозольный путь : Синтез MeCbl, который служит коферментом метионинсинтазы.
Когда оба пути нарушены из-за потери функции MMACHC:
Накопление MMA : Дефицит AdoCbl → снижение активности метилмалонил-КоА-мутазы → накопление MMA → нейротоксичность (метаболический инсульт)
Накопление Hcy : Дефицит MeCbl → снижение активности метионинсинтазы → накопление Hcy → повреждение сосудов и тромбоз2)·почечная ТМА2)
Снижение метионина : Нарушение превращения Hcy в метионин → снижение S-аденозилметионина (SAM) → нарушение метилирования → атрофия зрительного нерва
Что касается воздействия на ткани глаза, считается, что снижение синтеза глутатиона (GSH) из-за низкого уровня метионина ослабляет защиту от окислительного стресса пигментного эпителия сетчатки (ПЭС), способствуя макулярной дегенерации. Также предполагается механизм, при котором повреждение эндотелия сосудов из-за высокого уровня Hcy нарушает кровообращение сетчатки и хориоидеи.
7. Новейшие исследования и перспективы на будущее (отчеты на стадии исследований)
Hjalmarssон и соавт. (2024) сообщили о первом в мире случае имплантации LVAD (устройство вспомогательного левого желудочка) с последующей трансплантацией сердца у пациента с cblC и кардиомиопатией1). В сочетании с метаболической терапией высокими дозами OH-Cbl (30 мг/сут в/в), бетаином, фолиновой кислотой и L-карнитином были достигнуты улучшение метаболических показателей (MMA 97→8,1, Hcy 91→31,8) и хороший исход.
Было показано, что продолжение метаболической терапии cblC необходимо даже после трансплантации сердца1).
Ограничения неонатального скрининга и улучшение диагностики поздних форм
Было показано, что поздние и легкие формы могут быть пропущены при неонатальном скрининге4), и подчеркивается важность измерения Hcy и MMA у молодых взрослых с неврологическими или психиатрическими симптомами, даже при нормальном уровне B123, 4).
Сообщается об «epi-cblC», возникающем в результате эпигенетического сайленсинга MMACHC из-за мутации гена PRDX1. Даже при отсутствии мутации гена может проявляться фенотип, подобный cblC, что требует пересмотра диагностического алгоритма.
Ailliet и соавт. (2022) сообщили об опыте раннего выявления бессимптомного гомозигота при семейном скрининге позднего cblC, подчеркнув важность активного семейного скрининга после диагностики пробанда 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.