Le déficit en cobalamine C (type cblC) est une maladie congénitale du métabolisme intracellulaire de la vitamine B12 (cobalamine). Son nom complet est acidurie méthylmalonique et homocystinurie de type cblC (OMIM #277400). C’est le type le plus fréquent, représentant environ 80 % de tous les troubles du métabolisme de la cbl.
Le mode de transmission est autosomique récessif. La cause est une mutation du gène MMACHC (1p34.2). La protéine MMACHC est impliquée dans les premières étapes du métabolisme intracellulaire de la cobalamine ; sa perte de fonction entraîne un défaut de synthèse à la fois de l’adénosylcobalamine (AdoCbl) et de la méthylcobalamine (MeCbl).
L’incidence estimée est de 1:100 000 à 1:200 000 3, 4), mais une étude dans la province chinoise du Shandong a rapporté une fréquence élevée de 1:3 920. Aux États-Unis, cette maladie fait l’objet d’un dépistage néonatal (NBS) depuis le début des années 2000 4).
Selon l’âge de début, on distingue la forme à début précoce (avant 1 an, 86 à 88 % des cas) et la forme tardive (après 1 an). Plus de 160 cas de forme tardive et plus de 30 cas de début à l’âge adulte (≥18 ans) ont été rapportés 3).
QLe déficit en cobalamine C peut-il être détecté par le dépistage néonatal ?
A
Un dépistage néonatal basé sur un taux élevé de propionylcarnitine (C3) dans le sang permet une détection précoce. Cependant, les formes tardives ou légères peuvent passer inaperçues lors du dépistage 4) ; en cas de suspicion clinique malgré un dépistage normal, des examens complémentaires (Hcy plasmatique, MMA urinaire) sont nécessaires.
Le tableau clinique varie considérablement selon l’âge d’apparition.
Symptômes oculaires de la forme précoce :
Nystagmus : signe oculaire le plus précoce. Observé chez environ 70 à 76 % des patients.
Baisse de l’acuité visuelle : progresse dès la petite enfance et, même avec un traitement approprié, conduit souvent à la cécité légale à l’adolescence.
Angiographie en fluorescence du fond d’œil (FAF) : hypo-autofluorescence fovéolaire et hyper-autofluorescence en bordure.
Forme tardive
Fréquence de fond d’œil normal : souvent sans signes oculaires sévères3, 4).
IRM médullaire : lésions hyperintenses des cordons postérieurs caractéristiques4).
Lésions de la substance blanche à l’IRM cérébrale : observées dans certaines formes tardives3).
Rétinopathie hypertensive : peut survenir dans les cas graves avec complications rénales (microangiopathie thrombotique)2).
Les complications systémiques incluent acidose métabolique, hypotonie, microcéphalie, cardiomyopathie (50%)1) et microangiopathie thrombotique rénale (MAT)2).
QDes complications oculaires surviennent-elles aussi dans la forme tardive ?
A
Dans la forme tardive, la maculopathie sévère et la dégénérescence rétinienne de la forme précoce sont rares, et le fond d’œil est souvent normal3, 4). Cependant, dans les cas graves avec complications rénales, une rétinopathie hypertensive peut survenir2). L’amélioration des symptômes neurologiques répond souvent bien au traitement, mais des cas de déficience visuelle résiduelle ont été rapportés3).
La carence en cobalamine C est une maladie autosomique récessive due à des mutations du gène MMACHC. Plus de 80 mutations ont été rapportées, et le phénotype varie considérablement selon le type de mutation.
La fréquence des principales mutations et les phénotypes correspondants sont présentés ci-dessous.
Mutation
Fréquence / Lignée
Principal phénotype
c.271dupA
40–61 % chez les Européens
Apparition précoce, forme la plus sévère
c.394C>T
Environ 20 %
Forme tardive
c.609G>A
48–55 % chez les Asiatiques de l’Est
Nourrisson à enfant
Autres mutations principales :
c.331C>T : 5–9 % chez les Canadiens français1).
c.566G>A : associée à la forme tardive3).
c.484G>T : associée à une atteinte multiviscérale du nourrisson2).
c.271dupA + c.449T>A (hétérozygote composite) : des cas de forme tardive ont été rapportés4).
Il existe également un « epi-cblC » causé par un silencing épigénétique de MMACHC dû à une mutation du gène PRDX1, entraînant un phénotype cblC secondaire.
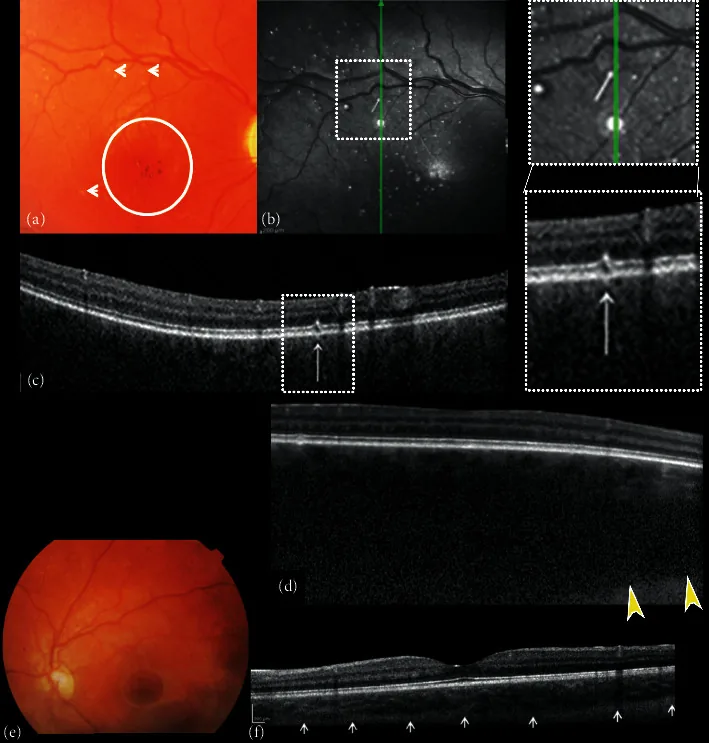
Composite de photographies couleur du fond d’œil et tomographie par cohérence optique en domaine spectral (SDOCT) des yeux droit et gauche. (a) Le fond d’œil droit montre une excavation et une pâleur du disque optique, absence de tessellation, hémangiome choroïdien diffus, hypo-hyperpigmentation de la zone fovéale avec réflexe fovéal absent (cercle), et de petites altérations en points blancs en forme de « microdrusen » (flèches). (b) Réflectance infrarouge proche (NIR) de l’œil droit montrant de multiples points hyperréfléchissants entourés d’un anneau hyporefléchissant correspondant aux petites altérations en points blancs en forme de « microdrusen » du pôle postérieur observées à l’ophtalmoscopie. La coupe B transversale SDOCT (c) sur les points hyperréfléchissants montre des altérations focales de la couche des photorécepteurs de l’épithélium pigmentaire rétinien (RPE).
Dépistage néonatal (NBS) : L’augmentation de la propionylcarnitine C3 par spectrométrie de masse en tandem est un indicateur. Cependant, les formes légères et tardives peuvent être manquées 4).
Les résultats de laboratoire montrant des anomalies métaboliques sont au cœur du diagnostic.
Paramètre
Résultat typique
Signification
Hcy plasmatique
Élevé de façon marquée
Dépistage
MMA urinaire
Élevé
Confirmation du bloc métabolique
Test génétique
Mutation MMACHC
Diagnostic de certitude
Valeurs numériques rapportées : Hcy plasmatique 101,5–1700,5 µmol/L2, 3), MMA urinaire 85–802 mmol/mol3, 4). La vitamine B12 sérique est souvent normale dans les formes tardives (448–625 pg/mL)3, 4).
Il est important de distinguer la carence alimentaire en B12, l’anémie pernicieuse, les autres anomalies du métabolisme des cobalamines (cblD, cblF, etc.) et le déficit en MTHFR3, 4). L’association d’une Hcy élevée et d’une MMA élevée est caractéristique du cblC, tandis qu’une Hcy élevée seule (MMA normale) suggère un déficit en MTHFR, etc.
QEst-il possible d'avoir un cblC alors que la vitamine B12 sérique est normale ?
A
Oui. Dans le cblC à début tardif, la B12 sérique est souvent dans la plage normale3, 4) ; un taux normal de B12 ne permet pas d’exclure le diagnostic. Chez les jeunes patients présentant des symptômes neurologiques ou psychiatriques, il est nécessaire de mesurer l’Hcy et la MMA plasmatiques, et si elles sont élevées, d’analyser le gène MMACHC.
Le traitement principal est l’administration parentérale d’hydroxocobalamine (OH-Cbl), associée à une polythérapie comprenant la bétaïne, l’acide folinique et la L-carnitine.
L’administration parentérale est la règle. Dose indicative : 1 mg/jour ou 0,3 mg/kg/jour en injection intramusculaire (IM)4). Doses rapportées :
Ailliet et al. (2022) ont administré 25 mg/jour en injection sous-cutanée (SC) à un adulte à début tardif, avec amélioration des symptômes neurologiques3). Goyne et al. (2023) ont débuté à 5 mg/jour IM4). Hjalmarsson et al. (2024) ont administré 30 mg/jour par voie intraveineuse (IV) chez un patient avec insuffisance cardiaque sévère1).
Favorise la reméthylation de l’homocystéine en méthionine. Dose indicative : 250 mg/kg/jour en 3 prises4). Doses rapportées : 100 mg/kg/jour2), 1 g × 3 fois4), 2-3 g × 3 fois1).
Les objectifs du traitement sont la réduction de l’MMA et de l’Hcy et la normalisation des indicateurs métaboliques. Un traitement approprié permet une diminution significative de l’Hcy :
Akar et al. (2024) rapportent un cas sévère avec TMA rénale où l’Hcy est passée de 1 700,5 à 4,6 µmol/L après traitement2). Hjalmarssон et al. (2024) décrivent un cas avec cardiomyopathie où l’MMA est passée de 97 à 8,1 mmol/mol et l’Hcy de 91 à 31,8 µmol/L1).
Dans les formes tardives, la réponse au traitement est relativement bonne4), mais la récupération de la fonction visuelle peut être limitée3). Dans les formes précoces, même avec un traitement approprié, de nombreux patients atteignent la cécité légale à l’adolescence.
Cœur : Traitement de l’insuffisance cardiaque avec des bêta-bloquants, etc. Dans les cas graves, un LVAD (dispositif d’assistance ventriculaire gauche) ou une transplantation cardiaque peut être envisagé1).
Reins : Protection rénale par inhibiteurs de l’ECA ou ARA2).
QLes symptômes oculaires s'améliorent-ils avec le traitement ?
A
Dans les formes précoces, même si les indicateurs métaboliques s’améliorent, le pronostic visuel de la maculopathie et de la dégénérescence rétinienne est souvent mauvais, et des cas de cécité légale à l’adolescence sont rapportés. Dans les formes tardives, la réponse des symptômes neurologiques au traitement est relativement bonne, mais les troubles visuels résiduels ne se rétablissent que de manière limitée3). Un diagnostic et un traitement précoces pourraient offrir le meilleur pronostic.
La protéine MMACHC se lie au complexe cobalamine-haptocorrine (R) dans le lysosome, clive le groupe R de la cobalamine et génère un intermédiaire cobalamine commun utilisable dans la cellule. Cet intermédiaire se divise ensuite en deux voies.
Voie mitochondriale : Synthèse d’AdoCbl, qui agit comme coenzyme de la méthylmalonyl-CoA mutase.
Voie cytosolique : Synthèse de MeCbl, qui sert de coenzyme à la méthionine synthase.
Lorsque les deux voies sont perturbées par une perte de fonction de MMACHC :
Accumulation de MMA : Déficit en AdoCbl → activité réduite de la méthylmalonyl-CoA mutase → accumulation de MMA → neurotoxicité (accident métabolique)
Accumulation de Hcy : Déficit en MeCbl → activité réduite de la méthionine synthase → accumulation de Hcy → lésions vasculaires et thrombose2)·TMA rénale2)
Diminution de la méthionine : Conversion altérée de Hcy en méthionine → diminution de la S-adénosylméthionine (SAM) → trouble de la méthylation → atrophie optique
En ce qui concerne les tissus oculaires, on pense que la diminution de la synthèse du glutathion (GSH) due à un faible taux de méthionine réduit la défense contre le stress oxydatif de l’épithélium pigmentaire rétinien (EPR), contribuant ainsi à la dégénérescence maculaire. On suppose également que les lésions endothéliales vasculaires dues à un taux élevé de Hcy perturbent la circulation rétinienne et choroïdienne.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Hjalmarssон et al. (2024) ont rapporté le premier cas mondial d’implantation d’un LVAD (dispositif d’assistance ventriculaire gauche) suivie d’une transplantation cardiaque chez un patient cblC atteint de cardiomyopathie1). Associé à un traitement métabolique comprenant de l’OH-Cbl à haute dose (30 mg/jour IV), de la bétaïne, de l’acide folinique et de la L-carnitine, une amélioration des indicateurs métaboliques (MMA 97→8,1, Hcy 91→31,8) et un bon résultat ont été obtenus.
Il a été démontré que la poursuite du traitement métabolique pour cblC est essentielle même après la transplantation cardiaque1).
Limites du dépistage néonatal et amélioration du diagnostic des formes tardives
Il a été montré que certaines formes tardives et légères peuvent être manquées par le dépistage néonatal4), soulignant l’importance de mesurer Hcy et MMA chez les jeunes adultes présentant des symptômes neurologiques ou psychiatriques, même si la B12 est normale3, 4).
L’« epi-cblC » résultant du silençage épigénétique de MMACHC par une mutation du gène PRDX1 a été rapporté. Même en l’absence de mutation génétique, un phénotype de type cblC peut se manifester, ce qui nécessite de reconsidérer l’algorithme diagnostique.
Ailliet et al. (2022) ont rapporté l’expérience de la détection précoce d’un homozygote asymptomatique par dépistage familial dans un cas de cblC tardif, soulignant l’importance d’un dépistage familial actif suite au diagnostic du probant 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.