ภาวะขาดโคบาลามินซี (cblC) เป็นความผิดปกติของเมแทบอลิซึมวิตามินบี12ภายในเซลล์ที่เกิดจากการกลายพันธุ์ของยีน MMACHC ซึ่งเป็นชนิดที่พบบ่อยที่สุดในความผิดปกติของเมแทบอลิซึมโคบาลามินทั้งหมด คิดเป็นประมาณ 80% ของผู้ป่วย
ในชนิดที่เริ่มมีอาการเร็ว (ภายในปีแรกของชีวิต) จะเกิดจอประสาทตา เสื่อมและจอตาเสื่อมตั้งแต่ทารก และมักทำให้ตาบอดตามกฎหมายในวัยรุ่น
ในชนิดที่เริ่มมีอาการช้า (หลังอายุ 1 ปี) อาการทางระบบประสาทจะเด่นชัด และผลการตรวจอวัยวะภายในตามักไม่รุนแรงนัก
ระดับโฮโมซิสเทอีนในพลาสมา (Hcy) สูงขึ้นอย่างชัดเจน และกรดเมทิลมาโลนิกในปัสสาวะ (MMA) สูงขึ้นเป็นกุญแจสำคัญในการวินิจฉัย
แม้ว่าวิตามินบี 12 ในซีรัมจะปกติ ก็ไม่สามารถตัด cblC ออกได้ ซึ่งเป็นจุดหลอกลวงในการวินิจฉัย
การรักษาหลักคือการให้ไฮดรอกซีโคบาลามิน (OH-Cbl) ทางหลอดเลือดร่วมกับการให้เบทาอีนทางปาก
การพยากรณ์โรคด้านการมองเห็น ในชนิดที่เริ่มมีอาการเร็วมักไม่ดี ในขณะที่ชนิดที่เริ่มมีอาการช้าตอบสนองต่อการรักษาค่อนข้างดี
ภาวะขาดโคบาลามินซี (ชนิด cblC) เป็นโรคความผิดปกติแต่กำเนิดของเมแทบอลิซึมของวิตามินบี 12 (โคบาลามิน) ภายในเซลล์ ชื่อทางการคือ เมทิลมาโลนิกแอซิดูเรียและโฮโมซิสทินูเรียชนิด cblC (OMIM #277400) เป็นชนิดที่พบบ่อยที่สุด คิดเป็นประมาณ 80% ของความผิดปกติของเมแทบอลิซึมของ cbl ทั้งหมด
รูปแบบการถ่ายทอดทางพันธุกรรมเป็นแบบออโตโซมัลด้อย สาเหตุเกิดจากการกลายพันธุ์ของยีน MMACHC (1p34.2) โปรตีน MMACHC มีหน้าที่ในระยะเริ่มต้นของเมแทบอลิซึมของโคบาลามินภายในเซลล์ และการสูญเสียการทำงานของมันทำให้การสังเคราะห์ทั้งอะดีโนซิลโคบาลามิน (AdoCbl) และเมทิลโคบาลามิน (MeCbl) บกพร่อง
อัตราการเกิดโดยประมาณคือ 1:100,000 ถึง 1:200,000 3, 4) แต่การศึกษาในมณฑลซานตง ประเทศจีน รายงานความถี่สูงถึง 1:3,920 ในสหรัฐอเมริกา การตรวจคัดกรองทารกแรกเกิด (NBS) ได้กลายเป็นเป้าหมายตั้งแต่ต้นทศวรรษ 2000 4)
ตามอายุที่เริ่มมีอาการ แบ่งเป็น ชนิดเริ่มมีอาการเร็ว (ภายในปีแรกของชีวิต คิดเป็น 86-88% ของทั้งหมด) และ ชนิดเริ่มมีอาการช้า (หลังอายุ 1 ปี) มีรายงานผู้ป่วยชนิดเริ่มมีอาการช้ามากกว่า 160 ราย และผู้ป่วยที่เริ่มมีอาการในผู้ใหญ่ (อายุมากกว่า 18 ปี) มากกว่า 30 ราย 3)
Q
การตรวจคัดกรองทารกแรกเกิดสามารถตรวจพบภาวะขาดโคบาลามินซีได้หรือไม่?
A
การตรวจพบตั้งแต่เนิ่นๆ สามารถทำได้ผ่าน NBS โดยใช้ระดับโพรพิโอนิลคาร์นิทีน (C3) ในเลือดที่สูงขึ้นเป็นตัวบ่งชี้ อย่างไรก็ตาม ผู้ป่วยชนิดเริ่มมีอาการช้าหรือชนิดไม่รุนแรงอาจรอดจากการตรวจ NBS 4) และหากสงสัยทางคลินิกแม้ว่าผล NBS ปกติ ก็จำเป็นต้องตรวจเพิ่มเติมของ Hcy ในพลาสมาและ MMA ในปัสสาวะ
ภาพทางคลินิกแตกต่างกันอย่างมากตามอายุที่เริ่มมีอาการ
อาการทางตาที่เริ่มมีอาการเร็ว:
อาตา (Nystagmus):การมองเห็น ลดลง:ตาเหล่ (Strabismus):อาตา
อาการหลักที่เริ่มมีอาการช้า (นอกตา):
ขาอ่อนแรงและชา: อาการจากความผิดปกติของไขสันหลังส่วนหลัง (posterior column)4) ความผิดปกติของการเดินและอาการทางจิตเวช: มีรายงานในผู้ป่วยที่เริ่มมีอาการในวัยผู้ใหญ่3, 4)
ผลการตรวจอวัยวะภายในตา (fundus) แตกต่างกันอย่างมากระหว่างชนิดที่เริ่มมีอาการเร็วและช้า
ชนิดที่เริ่มมีอาการเร็ว
จอประสาทตา เสื่อม (Maculopathy):
จอประสาทตา เสื่อมแบบ Bull’s eye:จอประสาทตา และคอรอยด์ ฝ่อบริเวณศูนย์กลางร่วมกับวงแหวนเม็ดสี
การเปลี่ยนแปลงคล้ายจอประสาทตา อักเสบชนิดมีเม็ดสี (Retinitis pigmentosa-like changes): เกิดจอประสาทตา เสื่อมอย่างกว้างขวาง
ประสาทตาเสื่อม (Optic atrophy): พบในกรณีที่ดำเนินโรคมากแล้ว
การถ่ายภาพเรืองแสงของจอตา (FAF ) : รูปแบบการเรืองแสงอัตโนมัติต่ำที่รอยบุ๋มจอตา และการเรืองแสงอัตโนมัติสูงที่ขอบเขต
ชนิดเริ่มช้า
จอตามักปกติ : มักไม่พบความผิดปกติทางตาที่รุนแรง3, 4)
ผล MRI ไขสันหลัง : รอยโรคสัญญาณสูงที่คอลัมน์หลังเป็นลักษณะเฉพาะ4)
รอยโรคสารสีขาวในสมองจาก MRI : พบในบางกรณีของชนิดเริ่มช้า3)
จอประสาทตาเสื่อมจากความดันโลหิตสูง 2)
ภาวะแทรกซ้อนทั่วร่างกาย ได้แก่ ภาวะเลือดเป็นกรดจากการเผาผลาญ กล้ามเนื้ออ่อนแรง ศีรษะเล็ก กล้ามเนื้อหัวใจผิดปกติ (50%)1) และลิ่มเลือดอุดหลอดเลือดขนาดเล็กที่ไต (TMA)2)
Q
ชนิดเริ่มช้าทำให้เกิดภาวะแทรกซ้อนทางตาหรือไม่?
A
ในชนิดเริ่มช้า จอประสาทตา เสื่อมรุนแรงและจอตาเสื่อมเช่นชนิดเริ่มเร็วนั้นพบได้น้อย และจอตามักปกติ3, 4) อย่างไรก็ตาม ในกรณีรุนแรงที่มีภาวะแทรกซ้อนทางไต อาจเกิดจอประสาทตาเสื่อมจากความดันโลหิตสูง 2) อาการทางระบบประสาทมักตอบสนองต่อการรักษาได้ดี แต่มีรายงานผู้ป่วยที่ยังมีความบกพร่องทางการมองเห็น หลงเหลืออยู่3)
การขาดโคบาลามินซีเป็นโรคทางพันธุกรรมแบบถ่ายทอดทางออโตโซมด้อยจากการกลายพันธุ์ของยีน MMACHC มีรายงานการกลายพันธุ์มากกว่า 80 ชนิด และลักษณะฟีโนไทป์แตกต่างกันมากตามชนิดของการกลายพันธุ์
ความถี่ของการกลายพันธุ์หลักและฟีโนไทป์ที่สอดคล้องแสดงไว้ด้านล่าง
การกลายพันธุ์ ความถี่/สายพันธุ์ ฟีโนไทป์หลัก c.271dupA 40-61% · ชาวยุโรป เริ่มต้นเร็ว · รุนแรงที่สุด c.394C>T ประมาณ 20% ชนิดเริ่มช้า c.609G>A 48-55% · ชาวเอเชียตะวันออก ทารกถึงเด็ก
การกลายพันธุ์หลักอื่นๆ:
c.331C>T : 5-9% ในชาวแคนาดาเชื้อสายฝรั่งเศส1) .c.566G>A : เกี่ยวข้องกับชนิดเริ่มช้า3) .c.484G>T : เกี่ยวข้องกับความผิดปกติหลายอวัยวะในทารก2) .c.271dupA + c.449T>A (heterozygous แบบผสม) : มีรายงานกรณีที่แสดงอาการชนิดเริ่มช้า4) .
นอกจากนี้ยังมี “epi-cblC” ซึ่งเกิดจากการ silencing ทางเอพิเจเนติกส์ของ MMACHC เนื่องจากการกลายพันธุ์ของยีน PRDX1 ทำให้เกิดภาวะทุติยภูมิคล้าย cblC.
ภาวะขาดโคบาลามิน C เป็นโรคถ่ายทอดทางพันธุกรรมแบบด้อยบนออโตโซม พี่น้องของผู้ป่วยอาจเป็นพาหะของการกลายพันธุ์เดียวกัน การตรวจคัดกรองครอบครัวสามารถตรวจพบพาหะและผู้ป่วยที่ไม่มีอาการได้ตั้งแต่เนิ่นๆ ดังนั้นจึงแนะนำให้ปรึกษาแพทย์ผู้เชี่ยวชาญด้านพันธุศาสตร์
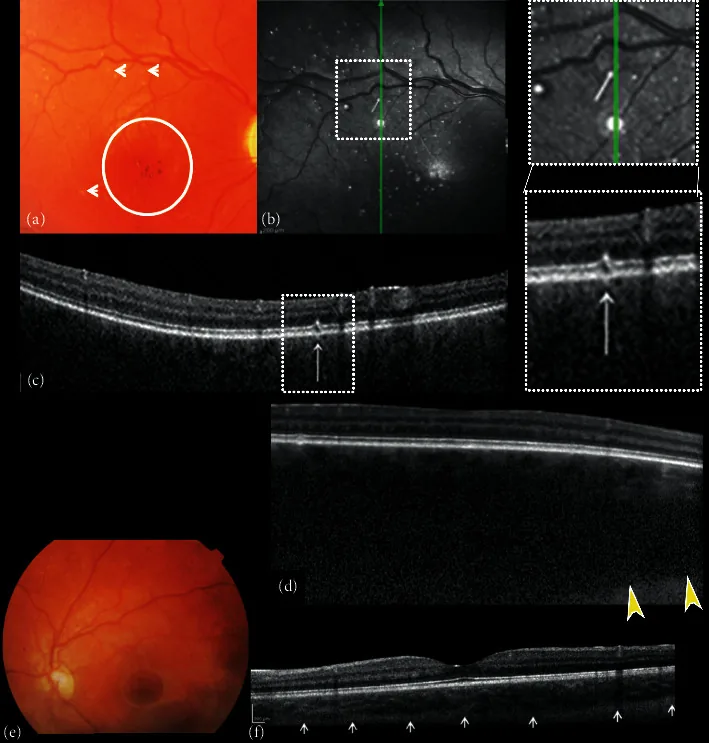
Solmaz Abdolrahimzadeh; Chiara Ciancimino; Flaminia Grassi; Edoardo Sordi; Serena Fragiotta; Gianluca Scuderi. Near-Infrared Reflectance Imaging in Retinal Diseases Affecting Young Patients. J Ophthalmol. 2021 Jul 31; 2021:5581851. Figure 4. PMCID: PMC8349282. License: CC BY.
ภาพประกอบจากภาพถ่ายสี
จอประสาทตา และการตรวจด้วยคลื่นแสงแบบสเปกตรัม (SDOCT) ของตาขวาและซ้าย (ก) ภาพ
จอประสาทตา ขวาแสดงการบุ๋มและสีซีดของ
จานประสาทตา ไม่มีลายเส้น หลอดเลือด
คอรอยด์ กระจาย การมีเม็ดสีมากเกินไปและน้อยเกินไปบริเวณโฟเวียโดยไม่มีรีเฟล็กซ์โฟเวีย (วงกลม) และการเปลี่ยนแปลงจุดสีขาวเล็กๆ คล้าย “ไมโคร
ดรูเซน ” (ลูกศร) (ข) การสะท้อนแสงอินฟราเรดใกล้ (NIR) ของตาขวาแสดง
จุดสะท้อนแสงสูง หลายจุดล้อมรอบด้วยวงแหวนสะท้อนแสงต่ำซึ่งสอดคล้องกับการเปลี่ยนแปลงจุดสีขาวเล็กๆ คล้าย “ไมโคร
ดรูเซน ” ที่ขั้วหลังซึ่งสังเกตได้จากการตรวจด้วยกล้องตรวจตา การสแกน B-scan SDOCT (ค) บน
จุดสะท้อนแสงสูง แสดงการเปลี่ยนแปลงเฉพาะจุดของชั้น
เยื่อบุผิวเม็ดสีจอประสาทตา (
RPE )-
เซลล์รับแสง
การตรวจคัดกรองทารกแรกเกิด (NBS) : การเพิ่มขึ้นของ C3 โพรพิโอนิลคาร์นิทีนโดยแมสสเปกโตรเมทรีแบบตีคู่เป็นตัวบ่งชี้ อย่างไรก็ตาม อาจพลาดกรณีที่ไม่รุนแรงและเริ่มมีอาการช้า 4)
ผลการตรวจที่แสดงความผิดปกติของเมแทบอลิซึมเป็นหัวใจสำคัญของการวินิจฉัย
รายการตรวจ ผลการตรวจทั่วไป ความสำคัญ Hcy ในพลาสมา สูงอย่างชัดเจน การคัดกรอง MMA ในปัสสาวะ สูง ยืนยันการอุดตันของเมแทบอลิซึม การตรวจทางพันธุกรรม การกลายพันธุ์ MMACHC การวินิจฉัยที่แน่นอน
ค่าตัวเลขที่รายงาน: Hcy ในพลาสมา 101.5–1700.5 µmol/L2, 3) , MMA ในปัสสาวะ 85–802 mmol/mol3, 4) วิตามินบี 12 ในซีรั่มมักปกติในชนิดที่เริ่มมีอาการช้า (448–625 pg/mL)3, 4) .
การตรวจทางพันธุกรรม : การระบุการกลายพันธุ์ MMACHC โดย NGS หรือการหาลำดับโดยตรง2, 3) .การวิเคราะห์การทำงานของไฟโบรบลาสต์ที่เพาะเลี้ยง : ยืนยันการลดลงของการผลิต MeCbl และ AdoCbl3) .
MRI สมอง : ยืนยันรอยโรคของสารสีขาว3) และรอยโรคสัญญาณสูงในคอลัมน์หลัง4) .การตรวจอวัยวะและการเรืองแสงอัตโนมัติของอวัยวะ : การประเมินรูปแบบของจอประสาทตา เสื่อม
สิ่งสำคัญคือต้องแยกความแตกต่างระหว่างการขาด B12 จากอาหาร โรคโลหิตจางชนิดเพอร์นิเชียส ความผิดปกติของการเผาผลาญ cbl อื่นๆ (เช่น cblD, cblF) และการขาด MTHFR3, 4) การรวมกันของ Hcy สูงและ MMA สูงเป็นลักษณะเฉพาะของ cblC ในขณะที่ Hcy สูงเพียงอย่างเดียว (MMA ปกติ) บ่งชี้ถึงการขาด MTHFR เป็นต้น
Q
เป็นไปได้หรือไม่ที่จะเป็น cblC แม้ว่าวิตามิน B12 ในซีรัมจะปกติ?
A
ได้ ใน cblC ที่เริ่มมีอาการช้า B12 ในซีรัมมักอยู่ในช่วงปกติ3, 4) และไม่ควรตัดออกโดยพิจารณาจาก B12 ปกติเพียงอย่างเดียว ในผู้ป่วยอายุน้อยที่มีอาการทางระบบประสาทหรือจิตเวช ควรวัด Hcy และ MMA ในพลาสมา และหากสูง จำเป็นต้องวิเคราะห์ยีน MMACHC
การรักษาหลักคือการให้ไฮดรอกซีโคบาลามิน (OH-Cbl) ทางหลอดเลือด ร่วมกับเบทาอีน กรดโฟลินิก และแอล-คาร์นิทีนในการรักษาแบบหลายยา
การให้ทางหลอดเลือดเป็นพื้นฐาน ขนาดยาโดยประมาณ: 1 มก./วัน หรือ 0.3 มก./กก./วัน ฉีดเข้ากล้าม (IM)4) ขนาดยาที่รายงาน:
ในผู้ใหญ่ที่เริ่มมีอาการช้า Ailliet และคณะ (2022) ให้ 25 มก./วัน ฉีดใต้ผิวหนัง (SC) และทำให้อาการทางระบบประสาทดีขึ้น3) ในกรณีของ Goyne และคณะ (2023) เริ่มด้วย 5 มก./วัน IM4) ในกรณีของ Hjalmarsson และคณะ (2024) ที่มีภาวะหัวใจล้มเหลวรุนแรง ให้ 30 มก./วัน ทางหลอดเลือดดำ (IV)1)
ส่งเสริมการรีเมทิลเลชันของโฮโมซิสเทอีนเป็นเมไทโอนีน ขนาดยาโดยประมาณ: 250 มก./กก./วัน แบ่งให้ 3 ครั้ง4) ขนาดยาที่รายงาน: 100 มก./กก./วัน2) , 1 กรัม × 3 ครั้ง4) , 2-3 กรัม × 3 ครั้ง1)
กรดโฟลินิก (ลิวโคโวริน) : 15-20 มก./วัน1) แอล-คาร์นิทีน : 2 กรัม × 2 ครั้ง1) , 660 มก. × 3 ครั้ง4)
เป้าหมายของการรักษาคือการลด MMA และ Hcy และทำให้ตัวชี้วัดทางเมตาบอลิกกลับสู่ปกติ การรักษาที่เหมาะสมจะทำให้ Hcy ลดลงอย่างชัดเจน:
ในกรณีรุนแรงที่มี TMA ของไตจาก Akar และคณะ (2024) Hcy กลับสู่ปกติจาก 1,700.5 เป็น 4.6 µmol/L หลังจากเริ่มการรักษา2) ในกรณีที่มีโรคกล้ามเนื้อหัวใจจาก Hjalmarsson และคณะ (2024) MMA ดีขึ้นจาก 97 เป็น 8.1 mmol/mol และ Hcy จาก 91 เป็น 31.8 µmol/L1)
ในชนิดที่เริ่มมีอาการช้า การตอบสนองต่อการรักษาค่อนข้างดี4) แต่การฟื้นฟูการมองเห็น อาจมีจำกัด3) ในชนิดที่เริ่มมีอาการเร็ว แม้จะได้รับการรักษาที่เหมาะสม ผู้ป่วยจำนวนมากถึงภาวะตาบอดตามกฎหมายในวัยรุ่น
หัวใจ : การรักษาภาวะหัวใจล้มเหลวด้วยยา beta-blocker เป็นต้น ในกรณีรุนแรง อาจพิจารณา LVAD (เครื่องช่วยหัวใจห้องล่างซ้าย) หรือการปลูกถ่ายหัวใจ1) ไต : การปกป้องไตด้วย ACE inhibitor หรือ ARB2)
ไนตรัสออกไซด์ (แก๊สหัวเราะ) ออกซิไดซ์วิตามินบี 12 อย่างไม่สามารถย้อนกลับได้ ดังนั้นจึงห้ามใช้ในผู้ป่วย cblC4) ต้องแจ้งแพทย์ผู้รักษาทุกครั้งก่อนทำฟันหรือการวางยาสลบ
ควรหลีกเลี่ยงการจำกัดโปรตีนมากเกินไปเพราะจะทำให้เมไทโอนีนลดลง4)
ในชนิดที่เริ่มมีอาการเร็ว การพยากรณ์โรคของการมองเห็น มักไม่ดีแม้จะได้รับการรักษา ดังนั้นการฟื้นฟูสมรรถภาพทางสายตาและการศึกษาสนับสนุนตั้งแต่เนิ่นๆ จึงมีความสำคัญ
Q
อาการทางตาดีขึ้นด้วยการรักษาหรือไม่?
A
ในชนิดที่เริ่มมีอาการเร็ว แม้ตัวชี้วัดทางเมตาบอลิกจะดีขึ้น การพยากรณ์โรคของการมองเห็น สำหรับโรคจุดภาพชัด และจอประสาทตา เสื่อมมักไม่ดี และมีรายงานกรณีที่ถึงภาวะตาบอดตามกฎหมายในวัยรุ่น ในชนิดที่เริ่มมีอาการช้า การตอบสนองของอาการทางระบบประสาทต่อการรักษาค่อนข้างดี แต่ความบกพร่องทางการมองเห็น ที่เหลืออยู่จะดีขึ้นอย่างจำกัดเท่านั้น3) การวินิจฉัยและการรักษาตั้งแต่เนิ่นๆ อาจให้การพยากรณ์โรคที่ดีที่สุด
โปรตีน MMACHC จับกับสารเชิงซ้อนโคบาลามิน-แฮปโตคอริน (R) ในไลโซโซม แยกหมู่ R ออกจากโคบาลามินเพื่อสร้างตัวกลางโคบาลามินทั่วไปที่เซลล์สามารถนำไปใช้ได้ ตัวกลางนี้จะแตกแขนงออกเป็นสองวิถีทาง
วิถีไมโตคอนเดรีย : สังเคราะห์ AdoCbl และทำหน้าที่เป็นโคเอนไซม์สำหรับเมทิลมาโลนิล CoA มิวเทสวิถีไซโตพลาสซึม : สังเคราะห์ MeCbl และทำหน้าที่เป็นโคเอนไซม์ของเมไธโอนีนซินเทส
เมื่อวิถีทั้งสองถูกรบกวนเนื่องจากการสูญเสียการทำงานของ MMACHC:
การสะสมของ MMA : การขาด AdoCbl → กิจกรรมของเมทิลมาโลนิล-CoA มิวเทสลดลง → การสะสมของ MMA → พิษต่อระบบประสาท (โรคหลอดเลือดสมองจากเมตาบอลิก)การสะสมของ Hcy : การขาด MeCbl → กิจกรรมของเมไธโอนีนซินเทสลดลง → การสะสมของ Hcy → ความเสียหายของหลอดเลือดและการเกิดลิ่มเลือด2) และ TMA ของไต2) ระดับเมไธโอนีนต่ำ : การเปลี่ยน Hcy เป็นเมไธโอนีนบกพร่อง → S-อะดีโนซิลเมไธโอนีน (SAM) ลดลง → เมทิลเลชันบกพร่อง → ฝ่อของเส้นประสาทตา
สำหรับผลกระทบต่อเนื้อเยื่อตา เชื่อว่าการสังเคราะห์กลูตาไธโอน (GSH) ที่บกพร่องเนื่องจากระดับเมไธโอนีนต่ำจะลดการป้องกันความเครียดออกซิเดชันของเยื่อบุผิวรงควัตถุจอตา (RPE ) ซึ่งเป็นสาเหตุหนึ่งของจอประสาทตา เสื่อม นอกจากนี้ยังสันนิษฐานว่าความเสียหายของเยื่อบุผนังหลอดเลือดจาก Hcy สูงจะรบกวนการไหลเวียนของจอตาและคอรอยด์
Hjalmarssон และคณะ (2024) รายงานผู้ป่วยรายแรกของโลกที่ได้รับการฝัง LVAD (เครื่องช่วยพยุงหัวใจห้องล่างซ้าย) ตามด้วยการปลูกถ่ายหัวใจในผู้ป่วย cblC ที่มีโรคกล้ามเนื้อหัวใจ1) ร่วมกับการรักษาทางเมตาบอลิกด้วย OH-Cbl ขนาดสูง (30 มก./วัน ทางหลอดเลือดดำ), เบทาอีน, กรดโฟลินิก และแอล-คาร์นิทีน ส่งผลให้ตัวชี้วัดทางเมตาบอลิกดีขึ้น (MMA 97→8.1, Hcy 91→31.8) และผลลัพธ์ที่ดี
กรณีนี้แสดงให้เห็นว่าการรักษาทางเมตาบอลิกสำหรับ cblC อย่างต่อเนื่องหลังการปลูกถ่ายหัวใจเป็นสิ่งจำเป็น1)
การศึกษาชี้ให้เห็นว่าผู้ป่วยบางรายชนิดที่เริ่มมีอาการช้าหรือไม่รุนแรงอาจถูกมองข้ามในการตรวจคัดกรองทารกแรกเกิด4) โดยเน้นย้ำถึงความสำคัญของการวัด Hcy และ MMA ในผู้ใหญ่วัยหนุ่มสาวที่มีอาการทางระบบประสาทหรือจิตเวช แม้ว่าระดับ B12 จะปกติ3, 4)
มีการรายงาน “epi-cblC” ที่เกิดจากการ silencing ระดับเอพิเจเนติกส์ของ MMACHC เนื่องจากการกลายพันธุ์ของยีน PRDX1 เนื่องจากสามารถแสดงฟีโนไทป์คล้าย cblC ได้แม้ไม่มีการกลายพันธุ์ของยีน จึงจำเป็นต้องพิจารณาทบทวนอัลกอริทึมการวินิจฉัย
Ailliet และคณะ (2022) รายงานประสบการณ์การตรวจพบ homozygous ที่ไม่มีอาการในระยะเริ่มต้นผ่านการตรวจคัดกรองครอบครัวใน cblC ชนิดเริ่มช้า และเน้นย้ำถึงความสำคัญของการตรวจคัดกรองครอบครัวเชิงรุกหลังการวินิจฉัย proband 3)
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.