Cobalamin C deficiency (cblC type) is an inborn error of intracellular vitamin B12 (cobalamin) metabolism. Its formal name is methylmalonic aciduria and homocystinuria, cblC type (OMIM #277400). It is the most common type, accounting for about 80% of all cbl metabolism disorders.
The inheritance pattern is autosomal recessive. The cause is mutations in the MMACHC gene (1p34.2). The MMACHC protein is responsible for the early steps of intracellular cobalamin metabolism, and its loss of function impairs the synthesis of both adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl).
The estimated incidence is 1:100,000 to 1:200,000 3, 4), but a survey in Shandong Province, China reported a high frequency of 1:3,920. In the United States, it has been included in newborn screening (NBS) since the early 2000s 4).
Based on age of onset, it is broadly divided into early-onset type (within 1 year of life, 86-88% of all cases) and late-onset type (after 1 year of age). Over 160 cases of late-onset type have been reported, including more than 30 adult-onset cases (age 18 or older) 3).
QCan cobalamin C deficiency be detected by newborn screening?
A
Early detection is possible by NBS using elevated blood C3 propionylcarnitine as an indicator. However, some late-onset and mild cases may be missed by NBS 4), so if clinical suspicion remains despite normal screening, additional tests for plasma Hcy and urinary MMA are necessary.
Fundus autofluorescence (FAF): Low autofluorescence at the fovea and hyperautofluorescence at the border.
Late-onset type
Many cases have normal fundus: Often no severe ocular findings are present 3, 4).
Spinal MRI findings: High-signal lesions in the posterior columns are characteristic 4).
Brain MRI white matter lesions: Seen in some late-onset cases 3).
Hypertensive retinopathy: May occur in severe cases with renal complications (thrombotic microangiopathy) 2).
Systemic complications include metabolic acidosis, hypotonia, microcephaly, cardiomyopathy (50%) 1), and renal thrombotic microangiopathy (TMA) 2).
QCan ocular complications occur in late-onset type?
A
In late-onset type, severe maculopathy and retinal degeneration as seen in early-onset type are rare, and many cases have normal fundus 3, 4). However, hypertensive retinopathy may occur in severe cases with renal complications 2). Neurological symptoms often respond well to treatment, but cases with residual visual dysfunction have been reported 3).
Cobalamin C deficiency is an autosomal recessive disorder caused by mutations in the MMACHC gene. Over 80 mutations have been reported, and the phenotype varies greatly depending on the type of mutation.
The frequency of major mutations and corresponding phenotypes are shown below.
Mutation
Frequency/Lineage
Main phenotype
c.271dupA
40–61% in European populations
Early onset, most severe
c.394C>T
Approximately 20%
Late-onset form
c.609G>A
48–55% in East Asian populations
Infancy to childhood
Other major variants include:
c.331C>T: 5–9% in French Canadian populations1).
c.566G>A: Associated with late-onset form3).
c.484G>T: Associated with infantile multi-organ disease2).
c.271dupA + c.449T>A (compound heterozygote): Cases with late-onset form have been reported4).
Additionally, there is ‘epi-cblC’ caused by epigenetic silencing of MMACHC due to PRDX1 gene mutations, leading to secondary cblC-like pathology.
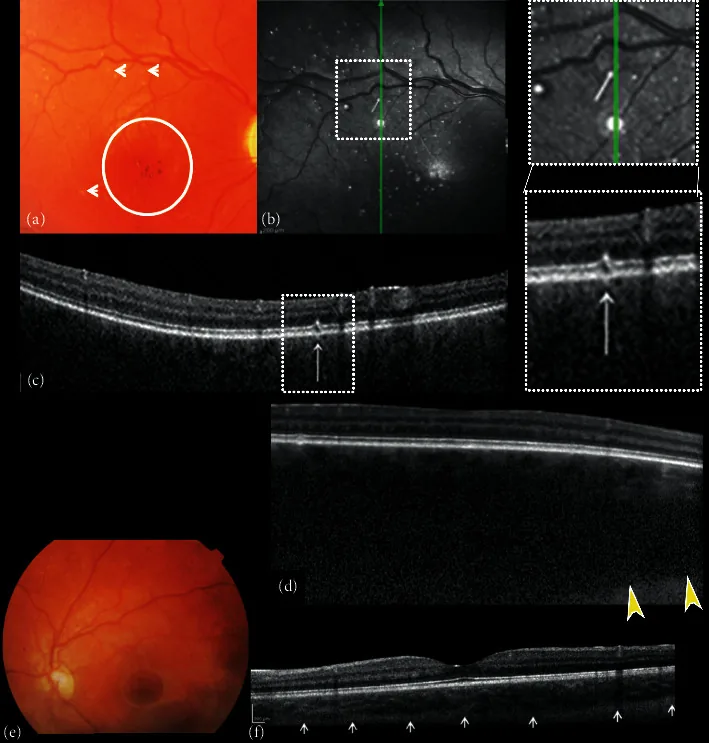
Composite of fundus color photographs and spectral-domain optical coherence (SDOCT) of the right and left eye. (a) The right fundus image shows excavation and pallor of the optic disk, absence of tessellation, diffuse choroidal hemangioma, hypo-hyper pigmentation of the foveal area with absent foveal reflex (circle), and small white dot shaped “microdrusen-like” alterations (arrows). (b) Near-infrared reflectance (NIR) of the right eye shows multiple hyperreflective dots surrounded by a hypo-reflective ring corresponding to the small white dot shaped “microdrusen-like” alterations of the posterior pole observed with ophthalmoscopy corresponded. B-scan cross-sectional SDOCT scan (c) on the hyperreflective dots shows focal alterations of the retinal pigment epithelial (RPE)-photoreceptor lay
Newborn screening (NBS): Elevated C3 propionylcarnitine by tandem mass spectrometry is an indicator. However, mild and late-onset forms may be missed 4).
Laboratory findings indicating metabolic abnormalities are central to diagnosis.
Test Item
Typical Findings
Significance
Plasma Hcy
Markedly elevated
Screening
Urinary MMA
Elevated
Confirms metabolic block
Genetic testing
MMACHC mutation
Definitive diagnosis
Reported specific values: plasma Hcy 101.5–1700.5 µmol/L2, 3), urinary MMA 85–802 mmol/mol3, 4). Serum vitamin B12 is often within normal range in late-onset forms (448–625 pg/mL)3, 4).
It is important to differentiate from dietary B12 deficiency, pernicious anemia, other cbl metabolism disorders (cblD, cblF, etc.), and MTHFR deficiency 3, 4). The combination of high Hcy and high MMA is characteristic of cblC, while high Hcy alone (normal MMA) suggests MTHFR deficiency, etc.
QIs it possible to have cblC even if serum vitamin B12 is normal?
A
Yes. In late-onset cblC, serum B12 is often within the normal range 3, 4), and it should not be ruled out based on normal B12 alone. In young patients with neurological or psychiatric symptoms, plasma Hcy and MMA must be measured, and if elevated, genetic testing for MMACHC should be performed.
The mainstay of treatment is parenteral administration of hydroxocobalamin (OH-Cbl), combined with betaine, folinic acid, and L-carnitine in a multi-drug regimen.
Parenteral administration is standard. Recommended dose: 1 mg/day or 0.3 mg/kg/day intramuscular injection (IM) 4). Reported doses:
Ailliet et al. (2022) administered 25 mg/day subcutaneous (SC) injection in a late-onset adult case, achieving neurological improvement 3). Goyne et al. (2023) started with 5 mg/day IM 4). Hjalmarsson et al. (2024) used 30 mg/day intravenous (IV) administration in a case complicated by severe heart failure 1).
Promotes remethylation of homocysteine to methionine. Recommended dose: 250 mg/kg/day divided into three doses 4). Reported doses: 100 mg/kg/day 2), 1 g three times daily 4), 2–3 g three times daily 1).
The treatment goals are reduction of MMA and Hcy and normalization of metabolic parameters. Appropriate treatment leads to a marked decrease in Hcy:
In a severe case with renal TMA reported by Akar et al. (2024), Hcy normalized from 1,700.5 to 4.6 µmol/L after treatment initiation 2). In a case with cardiomyopathy reported by Hjalmarsson et al. (2024), MMA improved from 97 to 8.1 mmol/mol and Hcy from 91 to 31.8 µmol/L 1).
In late-onset forms, treatment response is relatively good 4), but visual function recovery may be limited 3). In early-onset forms, even with appropriate treatment, many patients become legally blind in their teens.
Heart: Heart failure treatment with beta-blockers, etc. In severe cases, LVAD (left ventricular assist device) or heart transplantation may be considered 1).
Kidneys: Renal protection with ACE inhibitors or ARBs 2).
QDo eye symptoms improve with treatment?
A
In early-onset forms, even if metabolic parameters improve, the visual prognosis for maculopathy and retinal degeneration is often poor, with many cases progressing to legal blindness in the teens. In late-onset forms, neurological symptoms respond relatively well to treatment, but residual visual impairment improves only to a limited extent 3). Early diagnosis and early treatment initiation may offer the best prognosis.
The MMACHC protein binds to the cobalamin-haptocorrin (R) complex in lysosomes, cleaving the R group from cobalamin to generate a common cobalamin intermediate that can be used intracellularly. This intermediate then branches into two pathways.
Mitochondrial pathway: Synthesizes AdoCbl, which functions as a coenzyme for methylmalonyl-CoA mutase.
Cytosolic pathway: Synthesizes MeCbl and functions as a coenzyme for methionine synthase.
When both pathways are impaired due to loss of MMACHC function:
Methionine decrease: Impaired conversion of Hcy to methionine → decreased S-adenosylmethionine (SAM) → methylation defects → optic atrophy
Regarding effects on ocular tissues, impaired glutathione (GSH) synthesis due to low methionine is thought to reduce oxidative stress defense in the retinal pigment epithelium (RPE), contributing to macular degeneration. It is also speculated that high Hcy-induced vascular endothelial damage impairs retinal and choroidal circulation.
7. Latest Research and Future Perspectives (Investigational Reports)
Hjalmarssон et al. (2024) reported the world’s first case of LVAD (left ventricular assist device) implantation followed by heart transplantation in a cblC patient with cardiomyopathy1). Combined with metabolic therapy including high-dose OH-Cbl (30 mg/day IV), betaine, folinic acid, and L-carnitine, favorable outcomes were achieved with improvement in metabolic markers (MMA 97→8.1, Hcy 91→31.8).
It was shown that continuation of metabolic therapy for cblC is essential even after heart transplantation1).
Limitations of Newborn Screening and Improved Diagnosis of Late-Onset Forms
It has been shown that some late-onset and mild cases are missed by NBS4), emphasizing the importance of measuring Hcy and MMA even when B12 is normal in young adults presenting with neurological or psychiatric symptoms3, 4).
“epi-cblC” caused by epigenetic silencing of MMACHC due to PRDX1 gene mutation has been reported. Since cblC-like pathology can occur even without gene mutations, reconsideration of the diagnostic algorithm is required.
Ailliet et al. (2022) reported the early detection of asymptomatic homozygotes through family screening for late-onset cblC, emphasizing the significance of proactive family screening triggered by proband diagnosis 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.