La deficiencia de cobalamina C (tipo cblC) es un error congénito del metabolismo intracelular de la vitamina B12 (cobalamina). Su nombre formal es aciduria metilmalónica y homocistinuria tipo cblC (OMIM #277400). Es el tipo más común, representando aproximadamente el 80% de todos los trastornos del metabolismo de cbl.
El patrón de herencia es autosómico recesivo. La causa son mutaciones en el gen MMACHC (1p34.2). La proteína MMACHC es responsable de los pasos iniciales del metabolismo intracelular de la cobalamina, y su pérdida de función afecta la síntesis tanto de adenosilcobalamina (AdoCbl) como de metilcobalamina (MeCbl).
La incidencia estimada es de 1:100,000 a 1:200,000 3, 4), pero una encuesta en la provincia de Shandong, China, reportó una alta frecuencia de 1:3,920. En los Estados Unidos, se ha incluido en el cribado neonatal (NBS) desde principios de la década de 2000 4).
Según la edad de inicio, se divide ampliamente en tipo de inicio temprano (dentro del primer año de vida, 86-88% de todos los casos) y tipo de inicio tardío (después del año de edad). Se han reportado más de 160 casos de inicio tardío, incluyendo más de 30 casos de inicio en adultos (18 años o más) 3).
Q¿Se puede detectar la deficiencia de cobalamina C mediante el cribado neonatal?
A
La detección temprana es posible mediante NBS utilizando la elevación de C3 propionilcarnitina en sangre como indicador. Sin embargo, algunos casos de inicio tardío y leves pueden pasar desapercibidos en el NBS 4), por lo que si hay sospecha clínica a pesar de un cribado normal, son necesarias pruebas adicionales de Hcy plasmática y MMA urinaria.
Muchos casos tienen fondo de ojo normal: A menudo no presentan hallazgos oculares graves 3, 4).
Hallazgos en RM espinal: Lesiones hiperintensas en los cordones posteriores son características 4).
Lesiones de sustancia blanca en RM cerebral: Se observan en algunos casos de inicio tardío 3).
Retinopatía hipertensiva: Puede ocurrir en casos graves con complicaciones renales (microangiopatía trombótica) 2).
Las complicaciones sistémicas incluyen acidosis metabólica, hipotonía, microcefalia, miocardiopatía (50%) 1) y microangiopatía trombótica renal (TMA) 2).
Q¿Pueden ocurrir complicaciones oculares en el tipo de inicio tardío?
A
En el tipo de inicio tardío, la maculopatía grave y la degeneración retiniana como en el tipo de inicio temprano son raras, y muchos casos tienen fondo de ojo normal 3, 4). Sin embargo, puede presentarse retinopatía hipertensiva en casos graves con complicaciones renales 2). Los síntomas neurológicos suelen responder bien al tratamiento, pero se han reportado casos con deterioro visual residual 3).
La deficiencia de cobalamina C es un trastorno autosómico recesivo causado por mutaciones en el gen MMACHC. Se han reportado más de 80 mutaciones, y el fenotipo varía mucho según el tipo de mutación.
La frecuencia de las mutaciones principales y los fenotipos correspondientes se muestran a continuación.
Mutación
Frecuencia/Linaje
Fenotipo principal
c.271dupA
40–61% en poblaciones europeas
Inicio temprano, más grave
c.394C>T
Aproximadamente 20%
Forma de inicio tardío
c.609G>A
48–55% en poblaciones de Asia oriental
Lactancia a niñez
Otras variantes principales incluyen:
c.331C>T: 5–9% en poblaciones francocanadienses1).
c.566G>A: Asociada con la forma de inicio tardío3).
c.484G>T: Asociada con enfermedad multiorgánica infantil2).
c.271dupA + c.449T>A (heterocigoto compuesto): Se han reportado casos con forma de inicio tardío4).
Además, existe ‘epi-cblC’ causado por silenciamiento epigenético de MMACHC debido a mutaciones en el gen PRDX1, lo que provoca una patología secundaria similar a cblC.
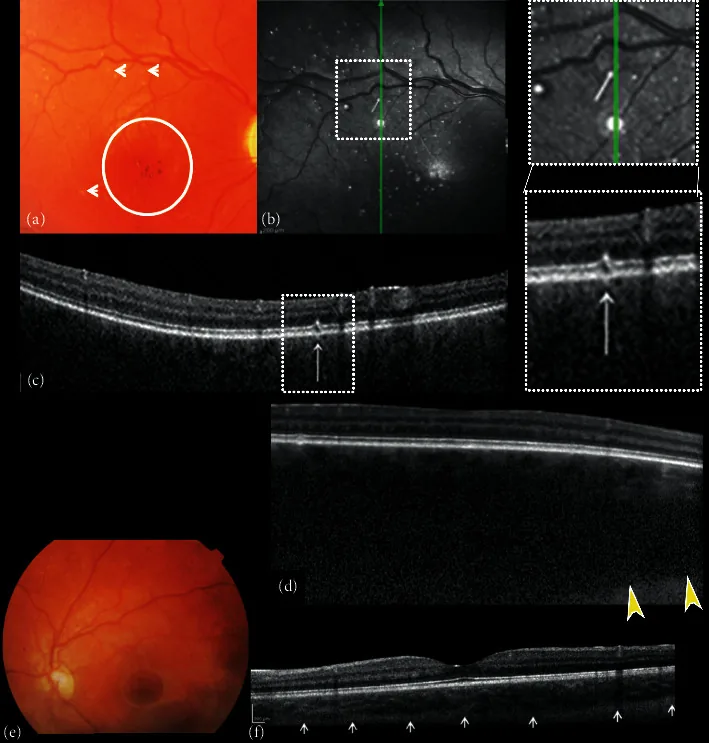
Composición de fotografías de fondo de ojo en color y tomografía de coherencia óptica de dominio espectral (SDOCT) del ojo derecho e izquierdo. (a) La imagen del fondo de ojo derecho muestra excavación y palidez del disco óptico, ausencia de teselación, hemangioma coroideo difuso, pigmentación hipo-hiper de la zona foveal con reflejo foveal ausente (círculo) y pequeñas alteraciones en forma de puntos blancos “similares a microdrusas” (flechas). (b) La reflectancia infrarroja cercana (NIR) del ojo derecho muestra múltiples puntos hiperreflectivos rodeados por un anillo hiporreflectivo que corresponden a las pequeñas alteraciones en forma de puntos blancos “similares a microdrusas” del polo posterior observadas con oftalmoscopia. La exploración SDOCT de corte transversal B-scan (c) sobre los puntos hiperreflectivos muestra alteraciones focales de la capa de fotorreceptores del epitelio pigmentario de la retina (RPE)
Cribado neonatal (NBS): La elevación de C3 propionilcarnitina por espectrometría de masas en tándem es un indicador. Sin embargo, las formas leves y de inicio tardío pueden pasarse por alto 4).
Los hallazgos de laboratorio que indican anomalías metabólicas son centrales para el diagnóstico.
Elemento de prueba
Hallazgos típicos
Significado
Hcy plasmática
Marcadamente elevada
Cribado
MMA urinario
Elevado
Confirma bloqueo metabólico
Prueba genética
Mutación MMACHC
Diagnóstico definitivo
Valores específicos reportados: Hcy plasmática 101.5–1700.5 µmol/L2, 3), MMA urinario 85–802 mmol/mol3, 4). La vitamina B12 sérica suele estar en rango normal en formas de inicio tardío (448–625 pg/mL)3, 4).
Es importante diferenciar de la deficiencia dietética de B12, anemia perniciosa, otros trastornos del metabolismo de cbl (cblD, cblF, etc.) y deficiencia de MTHFR 3, 4). La combinación de Hcy elevado y MMA elevado es característica de cblC, mientras que solo Hcy elevado (MMA normal) sugiere deficiencia de MTHFR, etc.
Q¿Es posible tener cblC incluso si la vitamina B12 sérica es normal?
A
Sí. En cblC de inicio tardío, la B12 sérica suele estar dentro del rango normal 3, 4), y no debe descartarse solo por B12 normal. En pacientes jóvenes con síntomas neurológicos o psiquiátricos, se deben medir Hcy y MMA en plasma, y si están elevados, se debe realizar un análisis genético de MMACHC.
El pilar del tratamiento es la administración parenteral de hidroxocobalamina (OH-Cbl), combinada con betaína, ácido folínico y L-carnitina en un régimen de múltiples fármacos.
La administración parenteral es estándar. Dosis recomendada: 1 mg/día o 0.3 mg/kg/día por inyección intramuscular (IM) 4). Dosis reportadas:
Ailliet et al. (2022) administraron 25 mg/día por inyección subcutánea (SC) en un caso adulto de inicio tardío, obteniendo mejoría neurológica 3). Goyne et al. (2023) iniciaron con 5 mg/día IM 4). Hjalmarsson et al. (2024) utilizaron 30 mg/día por vía intravenosa (IV) en un caso complicado con insuficiencia cardíaca grave 1).
Promueve la remetilación de homocisteína a metionina. Dosis recomendada: 250 mg/kg/día dividido en tres dosis 4). Dosis reportadas: 100 mg/kg/día 2), 1 g tres veces al día 4), 2–3 g tres veces al día 1).
Los objetivos del tratamiento son reducir la MMA y la Hcy y normalizar los parámetros metabólicos. El tratamiento adecuado produce una disminución marcada de la Hcy:
En un caso grave con TMA renal informado por Akar et al. (2024), la Hcy se normalizó de 1.700,5 a 4,6 µmol/L después de iniciar el tratamiento 2). En un caso con miocardiopatía informado por Hjalmarsson et al. (2024), la MMA mejoró de 97 a 8,1 mmol/mol y la Hcy de 91 a 31,8 µmol/L 1).
En las formas de inicio tardío, la respuesta al tratamiento es relativamente buena 4), pero la recuperación de la función visual puede ser limitada 3). En las formas de inicio temprano, incluso con el tratamiento adecuado, muchos pacientes quedan legalmente ciegos en la adolescencia.
Corazón: Tratamiento de la insuficiencia cardíaca con betabloqueantes, etc. En casos graves, se puede considerar un LVAD (dispositivo de asistencia ventricular izquierda) o un trasplante cardíaco 1).
Riñones: Protección renal con inhibidores de la ECA o ARA II 2).
Q¿Mejoran los síntomas oculares con el tratamiento?
A
En las formas de inicio temprano, incluso si los parámetros metabólicos mejoran, el pronóstico visual para la maculopatía y la degeneración retiniana suele ser malo, y se han informado casos que progresan a ceguera legal en la adolescencia. En las formas de inicio tardío, los síntomas neurológicos responden relativamente bien al tratamiento, pero la discapacidad visual residual mejora solo de forma limitada 3). El diagnóstico temprano y el inicio temprano del tratamiento pueden ofrecer el mejor pronóstico.
La proteína MMACHC se une al complejo cobalamina-haptocorrina (R) en los lisosomas, separando el grupo R de la cobalamina para generar un intermediario común de cobalamina que puede utilizarse intracelularmente. Este intermediario luego se ramifica en dos vías.
Vía mitocondrial: Sintetiza AdoCbl, que funciona como coenzima para la metilmalonil-CoA mutasa.
Vía citosólica: Sintetiza MeCbl y funciona como coenzima de la metionina sintasa.
Cuando ambas vías se ven afectadas por la pérdida de función de MMACHC:
Acumulación de MMA: Deficiencia de AdoCbl → disminución de la actividad de la metilmalonil-CoA mutasa → acumulación de MMA → neurotoxicidad (ictus metabólico)
Acumulación de Hcy: Deficiencia de MeCbl → disminución de la actividad de la metionina sintasa → acumulación de Hcy → daño vascular, trombosis2), TMA renal2)
Disminución de metionina: Alteración de la conversión de Hcy a metionina → disminución de S-adenosilmetionina (SAM) → defectos de metilación → atrofia óptica
En cuanto a los efectos en los tejidos oculares, se cree que la alteración de la síntesis de glutatión (GSH) debido a la baja metionina reduce la defensa contra el estrés oxidativo en el epitelio pigmentario de la retina (EPR), contribuyendo a la degeneración macular. También se especula que el daño endotelial vascular inducido por Hcy elevado afecta la circulación retiniana y coroidea.
7. Investigación más reciente y perspectivas futuras (informes en fase de investigación)
Hjalmarssон et al. (2024) reportaron el primer caso mundial de implantación de LVAD (dispositivo de asistencia ventricular izquierda) seguido de trasplante cardíaco en un paciente con cblC y miocardiopatía1). Combinado con terapia metabólica que incluye OH-Cbl en dosis altas (30 mg/día IV), betaína, ácido folínico y L-carnitina, se obtuvieron resultados favorables con mejora de los marcadores metabólicos (MMA 97→8.1, Hcy 91→31.8).
Se demostró que la continuación de la terapia metabólica para cblC es esencial incluso después del trasplante cardíaco1).
Limitaciones del cribado neonatal y mejora del diagnóstico de formas tardías
Se ha demostrado que algunos casos de inicio tardío y leves pasan desapercibidos en el cribado neonatal4), destacando la importancia de medir Hcy y MMA incluso con B12 normal en adultos jóvenes que presentan síntomas neurológicos o psiquiátricos3, 4).
Se ha reportado “epi-cblC” causado por el silenciamiento epigenético de MMACHC debido a una mutación en el gen PRDX1. Dado que puede presentarse una patología similar a cblC incluso sin mutaciones genéticas, se requiere reconsiderar el algoritmo diagnóstico.
Ailliet et al. (2022) reportaron la detección temprana de homocigotos asintomáticos mediante cribado familiar para cblC de inicio tardío, enfatizando la importancia del cribado familiar activo desencadenado por el diagnóstico del probando 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.