Il deficit di cobalamina C (tipo cblC) è una malattia congenita del metabolismo intracellulare della vitamina B12 (cobalamina). Il nome completo è aciduria metilmalonica e omocistinuria tipo cblC (OMIM #277400). È il tipo più frequente tra i disturbi del metabolismo della cbl, rappresentando circa l’80% dei casi.
La trasmissione è autosomica recessiva. La causa è una mutazione nel gene MMACHC (1p34.2). La proteina MMACHC è coinvolta nelle fasi iniziali del metabolismo intracellulare della cobalamina; la sua perdita di funzione porta a un difetto di sintesi sia dell’adenosilcobalamina (AdoCbl) che della metilcobalamina (MeCbl).
L’incidenza stimata è di 1:100.000-1:200.000 3, 4), ma uno studio nella provincia cinese dello Shandong ha riportato un’alta frequenza di 1:3.920. Negli Stati Uniti, questa malattia è oggetto di screening neonatale (NBS) dall’inizio degli anni 2000 4).
In base all’età di esordio, si distingue una forma a esordio precoce (entro il primo anno di vita, 86-88% dei casi) e una forma tardiva (dopo 1 anno). Sono stati riportati oltre 160 casi di forma tardiva e oltre 30 casi con esordio in età adulta (≥18 anni) 3).
QIl deficit di cobalamina C può essere rilevato dallo screening neonatale?
A
Un NBS basato su livelli elevati di C3-propionilcarnitina nel sangue consente una diagnosi precoce. Tuttavia, le forme tardive o lievi possono sfuggire allo screening 4); in caso di sospetto clinico nonostante uno screening normale, sono necessari ulteriori esami (Hcy plasmatico, MMA urinario).
Angiografia a fluorescenza del fondo oculare (FAF) : pattern di ipoautofluorescenza foveale e iperautofluorescenza al bordo.
Forma tardiva
Fondo oculare spesso normale : spesso senza segni oculari gravi3, 4).
RM midollare : lesioni iperintense dei cordoni posteriori caratteristiche4).
Lesioni della sostanza bianca alla RM cerebrale : osservate in alcune forme tardive3).
Retinopatia ipertensiva : può verificarsi in casi gravi con complicanze renali (microangiopatia trombotica)2).
Le complicanze sistemiche includono acidosi metabolica, ipotonia muscolare, microcefalia, cardiomiopatia (50%)1) e microangiopatia trombotica renale (MAT)2).
QSi verificano complicanze oculari anche nella forma tardiva?
A
Nella forma tardiva, la maculopatia grave e la degenerazione retinica della forma precoce sono rare, e il fondo oculare è spesso normale3, 4). Tuttavia, nei casi gravi con complicanze renali, può manifestarsi retinopatia ipertensiva2). I sintomi neurologici spesso rispondono bene al trattamento, ma sono stati riportati casi con deficit visivo residuo3).
La carenza di cobalamina C è una malattia autosomica recessiva causata da mutazioni nel gene MMACHC. Sono state riportate oltre 80 mutazioni e il fenotipo varia notevolmente a seconda del tipo di mutazione.
La frequenza delle mutazioni principali e i fenotipi corrispondenti sono mostrati di seguito.
Mutazione
Frequenza / Lignaggio
Fenotipo principale
c.271dupA
40–61% nei caucasici
Esordio precoce, forma più grave
c.394C>T
Circa il 20%
Forma tardiva
c.609G>A
48–55% negli asiatici orientali
Lattante–bambino
Altre mutazioni principali:
c.331C>T: 5–9% nei franco-canadesi1).
c.566G>A: associata alla forma tardiva3).
c.484G>T: associata a malattia multiorgano nel lattante2).
c.271dupA + c.449T>A (eterozigote composto): sono stati riportati casi con forma tardiva4).
Esiste anche un «epi-cblC» causato dal silenziamento epigenetico di MMACHC dovuto a una mutazione del gene PRDX1, che determina una condizione secondaria simile a cblC.
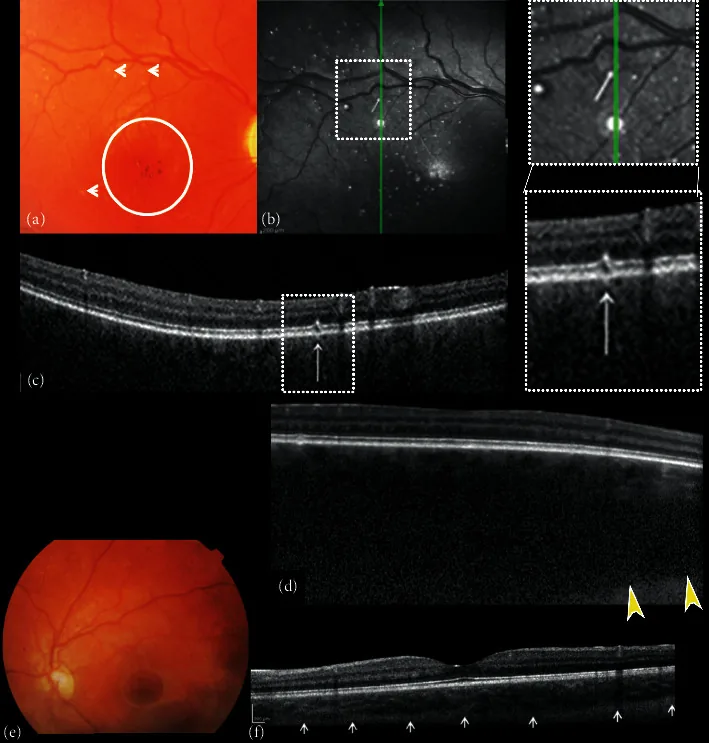
Composito di fotografie a colori del fondo e tomografia a coerenza ottica nel dominio spettrale (SDOCT) dell’occhio destro e sinistro. (a) L’immagine del fondo destro mostra escavazione e pallore del disco ottico, assenza di tassellazione, emangioma coroidale diffuso, ipo-iperpigmentazione dell’area foveale con riflesso foveale assente (cerchio) e piccole alterazioni a punti bianchi simili a “microdrusen” (frecce). (b) La riflettanza nel vicino infrarosso (NIR) dell’occhio destro mostra multipli punti iperriflettenti circondati da un anello iporiflettente corrispondenti alle piccole alterazioni a punti bianchi simili a “microdrusen” del polo posteriore osservate all’oftalmoscopia. La scansione B in sezione trasversale SDOCT (c) sui punti iperriflettenti mostra alterazioni focali dello strato dei fotorecettori dell’epitelio pigmentato retinico (RPE).
Screening neonatale (NBS) : L’aumento della propionilcarnitina C3 mediante spettrometria di massa tandem è un indicatore. Tuttavia, le forme lievi e a esordio tardivo possono essere trascurate 4).
È importante distinguere la carenza alimentare di B12, l’anemia perniciosa, altri disturbi del metabolismo delle cobalamine (cblD, cblF, ecc.) e il deficit di MTHFR3, 4). La combinazione di Hcy elevata e MMA elevata è caratteristica del cblC, mentre solo Hcy elevata (MMA normale) suggerisce un deficit di MTHFR, ecc.
QÈ possibile avere cblC nonostante la vitamina B12 sierica normale?
A
Sì. Nel cblC a esordio tardivo, la B12 sierica è spesso nell’intervallo normale3, 4); una B12 normale da sola non deve escludere la diagnosi. Nei giovani pazienti con sintomi neurologici o psichiatrici, è necessario dosare Hcy e MMA plasmatici; se elevati, è necessario analizzare il gene MMACHC.
Il cardine del trattamento è la somministrazione parenterale di idrossicobalamina (OH-Cbl), in combinazione con betaina, acido folinico e L-carnitina (polichemioterapia).
La somministrazione parenterale è la regola. Dose indicativa: 1 mg/die o 0,3 mg/kg/die per via intramuscolare (IM)4). Dosi riportate:
Ailliet et al. (2022) hanno somministrato 25 mg/die per via sottocutanea (SC) a un adulto a esordio tardivo, con miglioramento dei sintomi neurologici3). Goyne et al. (2023) hanno iniziato con 5 mg/die IM4). Hjalmarsson et al. (2024) hanno somministrato 30 mg/die per via endovenosa (EV) in un paziente con grave scompenso cardiaco1).
Favorisce la rimettilazione dell’omocisteina a metionina. Dose indicativa: 250 mg/kg/die in 3 dosi4). Dosi riportate: 100 mg/kg/die2), 1 g × 34), 2-3 g × 31).
Gli obiettivi del trattamento sono la riduzione di MMA e Hcy e la normalizzazione degli indicatori metabolici. Un trattamento appropriato porta a una marcata riduzione dell’Hcy:
Akar et al. (2024) riportano un caso grave con TMA renale in cui l’Hcy si è normalizzato da 1.700,5 a 4,6 µmol/L dopo l’inizio del trattamento2). Hjalmarssон et al. (2024) descrivono un caso con cardiomiopatia in cui MMA è migliorato da 97 a 8,1 mmol/mol e Hcy da 91 a 31,8 µmol/L1).
Nelle forme a esordio tardivo, la risposta al trattamento è relativamente buona4), ma il recupero della funzione visiva può essere limitato3). Nelle forme a esordio precoce, nonostante un trattamento adeguato, molti pazienti raggiungono la cecità legale nell’adolescenza.
Cuore: Trattamento dell’insufficienza cardiaca con beta-bloccanti, ecc. Nei casi gravi, si può considerare LVAD (dispositivo di assistenza ventricolare sinistra) o trapianto cardiaco1).
Reni: Protezione renale con ACE-inibitori o ARB2).
QI sintomi oculari migliorano con il trattamento?
A
Nelle forme a esordio precoce, anche se gli indicatori metabolici migliorano, la prognosi visiva per maculopatia e degenerazione retinica è spesso sfavorevole e sono riportati casi di cecità legale nell’adolescenza. Nelle forme a esordio tardivo, la risposta dei sintomi neurologici al trattamento è relativamente buona, ma il deficit visivo residuo si recupera solo in modo limitato3). Una diagnosi precoce e un trattamento tempestivo potrebbero offrire la migliore prognosi.
La proteina MMACHC si lega al complesso cobalamina-aptocorrina (R) nel lisosoma, scinde il gruppo R dalla cobalamina e genera un intermedio comune di cobalamina utilizzabile nella cellula. Questo intermedio si divide poi in due vie.
Via mitocondriale: Sintesi di AdoCbl, che funge da coenzima della metilmalonil-CoA mutasi.
Via citosolica : Sintesi di MeCbl, che funge da coenzima per la metionina sintasi.
Quando entrambe le vie sono compromesse dalla perdita di funzione di MMACHC:
Accumulo di MMA : Deficit di AdoCbl → ridotta attività della metilmalonil-CoA mutasi → accumulo di MMA → neurotossicità (ictus metabolico)
Accumulo di Hcy : Deficit di MeCbl → ridotta attività della metionina sintasi → accumulo di Hcy → danno vascolare e trombosi2)·TMA renale2)
Riduzione della metionina : Conversione alterata di Hcy in metionina → riduzione della S-adenosilmetionina (SAM) → alterazione della metilazione → atrofia ottica
Per quanto riguarda gli effetti sui tessuti oculari, si ritiene che la ridotta sintesi del glutatione (GSH) a causa del basso livello di metionina diminuisca la difesa dallo stress ossidativo dell’epitelio pigmentato retinico (RPE), contribuendo alla degenerazione maculare. Si ipotizza anche un meccanismo in cui il danno endoteliale vascolare dovuto all’alto Hcy compromette la circolazione retinica e coroidale.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Hjalmarssон et al. (2024) hanno riportato il primo caso mondiale di impianto di LVAD (dispositivo di assistenza ventricolare sinistra) seguito da trapianto cardiaco in un paziente cblC con cardiomiopatia1). In combinazione con terapia metabolica con OH-Cbl ad alte dosi (30 mg/die EV), betaina, acido folinico e L-carnitina, sono stati ottenuti un miglioramento degli indicatori metabolici (MMA 97→8,1, Hcy 91→31,8) e un buon esito.
È stato dimostrato che la continuazione della terapia metabolica per cblC è essenziale anche dopo il trapianto cardiaco1).
Limiti dello screening neonatale e miglioramento della diagnosi delle forme tardive
È stato dimostrato che le forme tardive e lievi possono essere trascurate dallo screening neonatale4), e viene sottolineata l’importanza di misurare Hcy e MMA nei giovani adulti con sintomi neurologici o psichiatrici, anche con B12 normale3, 4).
È stato riportato l’«epi-cblC» derivante dal silenziamento epigenetico di MMACHC a causa di una mutazione del gene PRDX1. Anche in assenza di mutazione genetica, può manifestarsi un fenotipo simile al cblC, richiedendo una riconsiderazione dell’algoritmo diagnostico.
Ailliet et al. (2022) hanno riportato l’esperienza di diagnosi precoce di un omozigote asintomatico tramite screening familiare in un caso di cblC a esordio tardivo, sottolineando l’importanza di uno screening familiare attivo dopo la diagnosi del probando 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.