早發型

鈷胺素C缺乏症
一目瞭然的要點
Section titled “一目瞭然的要點”1. 什麼是鈷胺素C缺乏症?
Section titled “1. 什麼是鈷胺素C缺乏症?”鈷胺素C缺乏症(cblC型)是一種細胞內維生素B12(鈷胺素)代謝的先天性代謝錯誤。正式名稱為甲基丙二酸尿症和高胱氨酸尿症cblC型(OMIM #277400)。它是所有cbl代謝異常中最常見的類型,約佔80%。
遺傳方式為體染色體隱性遺傳。病因是MMACHC基因(1p34.2)的突變。MMACHC蛋白負責細胞內鈷胺素代謝的早期步驟,其功能喪失會損害腺苷鈷胺素(AdoCbl)和甲基鈷胺素(MeCbl)的合成。
估計發生率為1:100,000至1:200,000 3, 4),但中國山東省的一項調查顯示高達1:3,920。在美國,自21世紀初以來,它已被納入新生兒篩檢(NBS)4)。
根據發病年齡,大致分為早發型(出生後1年內,佔所有病例的86-88%)和遲發型(1歲以後)。已報導超過160例遲發型病例,其中30例以上為成人發病(18歲及以上)3)。
Q
新生兒篩檢能發現鈷胺素C缺乏症嗎?
A
通過以血C3丙醯肉鹼升高為指標的NBS可以早期發現。然而,一些遲發型和輕症病例可能被NBS漏診4),因此如果臨床懷疑但篩檢正常,需要進一步檢查血漿Hcy和尿MMA。
2. 主要症狀和臨床表現
Section titled “2. 主要症狀和臨床表現”發病年齡不同,臨床表現差異很大。
早發型眼部症狀:
遲發型主要症狀(眼外):
- 下肢無力、麻木:脊髓後索功能障礙引起的症狀4)。
- 步行障礙、精神症狀:成人發病病例中有報導3, 4)。
早發型和遲發型的眼底所見差異很大。
晚發型
眼底正常者居多:常不呈現嚴重眼部表現 3, 4)。
脊髓MRI表現:後索高信號病變為特徵性表現 4)。
腦MRI白質病變:部分晚發型可見 3)。
高血壓性視網膜病變:在伴有腎臟併發症(血栓性微血管病)的重症病例中可能出現 2)。
全身併發症包括代謝性酸中毒、肌張力低下、小頭畸形、心肌病(50%)1)和腎血栓性微血管病(TMA)2)。
Q
晚發型也會發生眼部併發症嗎?
A
晚發型中,早發型那樣的嚴重黃斑病變和視網膜變性罕見,眼底正常者居多 3, 4)。但伴有腎臟併發症的重症病例可能出現高血壓性視網膜病變 2)。神經系統症狀通常對治療反應良好,但也有殘留視功能障礙的病例報告 3)。
3. 病因與風險因素
Section titled “3. 病因與風險因素”鈷胺素C缺乏症是由MMACHC基因突變引起的體染色體隱性遺傳病。已報導80多種突變,突變類型不同,表型差異很大。
主要突變的頻率及對應表型如下所示。
| 突變 | 頻率/譜系 | 主要表型 |
|---|---|---|
| c.271dupA | 40–61%·歐洲裔 | 早發型·最嚴重 |
| c.394C>T | 約20% | 晚發型 |
| c.609G>A | 48–55%·東亞裔 | 嬰兒至兒童 |
其他主要變異包括:
- c.331C>T:法裔加拿大人中占5–9%1)。
- c.566G>A:與晚發型相關3)。
- c.484G>T:與嬰兒期多器官疾病相關2)。
- c.271dupA + c.449T>A(複合雜合):已有晚發型病例報告4)。
此外,還存在由PRDX1基因突變導致MMACHC表觀遺傳沉默引起的「epi-cblC」,引發繼發性cblC樣病變。
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”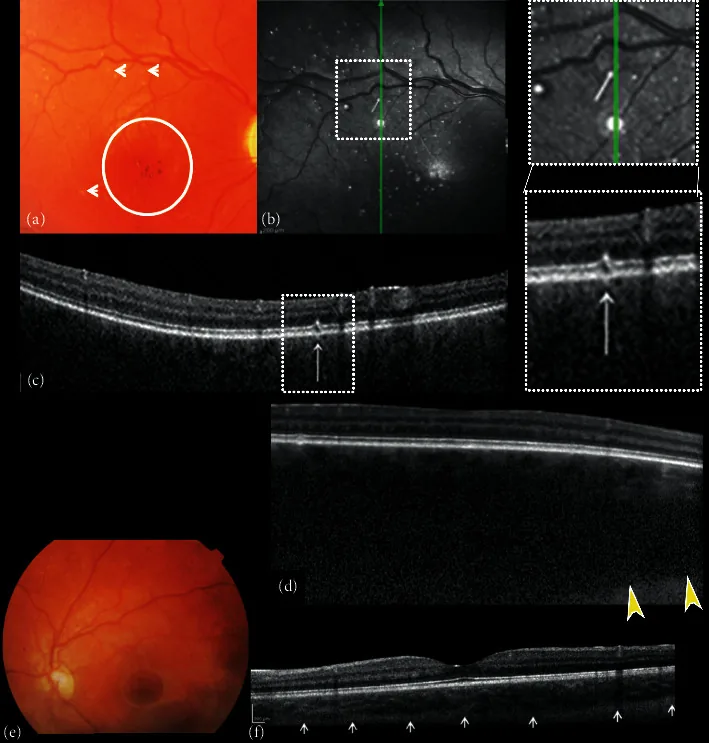
Solmaz Abdolrahimzadeh; Chiara Ciancimino; Flaminia Grassi; Edoardo Sordi; Serena Fragiotta; Gianluca Scuderi. Near-Infrared Reflectance Imaging in Retinal Diseases Affecting Young Patients. J Ophthalmol. 2021 Jul 31; 2021:5581851. Figure 4. PMCID: PMC8349282. License: CC BY.
右眼和左眼的眼底彩色照片和頻域光學相干斷層掃描(SDOCT)的合成圖。(a) 右眼底影像顯示視盤凹陷和蒼白,無脈絡膜紋理,瀰漫性脈絡膜血管瘤,中心凹區域色素沉著不均,中心凹反射消失(圓圈),以及小的白色點狀「微玻璃膜疣樣」改變(箭頭)。(b) 右眼的近紅外反射(NIR)顯示多個高反射點,周圍有低反射環,對應於眼底鏡檢查觀察到的後極部小白色點狀「微玻璃膜疣樣」改變。B掃描橫斷面SDOCT掃描(c)在高反射點上顯示視網膜色素上皮(RPE)-光感受器層的局部改變
- 新生兒篩檢(NBS):串聯質譜法檢測C3丙醯肉鹼升高為指標。但輕型和遲發型可能漏診4)。
血液和尿液檢查
Section titled “血液和尿液檢查”顯示代謝異常的實驗室檢查結果是診斷的核心。
| 檢查項目 | 典型表現 | 意義 |
|---|---|---|
| 血漿Hcy | 顯著升高 | 篩檢 |
| 尿MMA | 升高 | 確認代謝阻斷 |
| 基因檢測 | MMACHC突變 | 確診 |
報告的具體數值:血漿Hcy 101.5~1700.5 µmol/L2, 3),尿MMA 85~802 mmol/mol3, 4)。遲發型患者血清維生素B12常在正常範圍(448~625 pg/mL)3, 4)。
- 基因檢測:通過NGS或直接定序鑑定MMACHC突變2, 3)。
- 培養纖維母細胞功能分析:確認MeCbl和AdoCbl生成減少3)。
- 腦部MRI:顯示白質病變3)和後索高信號病變4)。
- 眼底檢查和眼底自發螢光:評估黃斑病變模式。
與飲食性B12缺乏、惡性貧血、其他cbl代謝異常(cblD、cblF等)以及MTHFR缺乏症的鑑別很重要3, 4)。高Hcy和高MMA的組合是cblC的特徵,而僅高Hcy(MMA正常)則提示MTHFR缺乏等。
Q
血清維生素B12正常時,是否可能患有cblC?
A
有可能。遲發型cblC患者的血清B12常在正常範圍內3, 4),不能僅憑B12正常就排除。對於有神經或精神症狀的年輕患者,必須檢測血漿Hcy和MMA,如果升高,需要進行MMACHC基因檢測。
5. 標準治療方法
Section titled “5. 標準治療方法”治療的核心是腸胃外給予羥鈷胺(OH-Cbl),並聯合使用甜菜鹼、亞葉酸和左卡尼汀的多藥聯合療法。
羥鈷胺(OH-Cbl)
Section titled “羥鈷胺(OH-Cbl)”腸胃外給藥是標準。推薦劑量:1 mg/天或0.3 mg/kg/天肌肉注射(IM)4)。報導的劑量:
Ailliet等人(2022)對一例遲發型成人患者給予25 mg/天皮下注射(SC),神經症狀得到改善3)。Goyne等人(2023)的病例起始劑量為5 mg/天IM4)。Hjalmarsson等人(2024)在一例合併嚴重心衰竭的病例中使用了30 mg/天靜脈注射(IV)1)。
促進同型半胱氨酸再甲基化為甲硫氨酸。推薦劑量:250 mg/kg/天,分三次服用4)。報導的劑量:100 mg/kg/天2),1 g每日三次4),2–3 g每日三次1)。
- 亞葉酸(甲醯四氫葉酸):15–20 mg/天1)。
- 左卡尼汀:2 g每日兩次1),660 mg每日三次4)。
治療目標與效果
Section titled “治療目標與效果”治療目標是降低MMA和Hcy,並使代謝指標正常化。適當治療可使Hcy顯著下降:
Akar等人(2024)報告的合併腎TMA的重症病例中,治療後Hcy從1,700.5 µmol/L降至4.6 µmol/L,恢復正常2)。Hjalmarsson等人(2024)報告的合併心肌病的病例中,MMA從97降至8.1 mmol/mol,Hcy從91降至31.8 µmol/L1)。
晚發型對治療反應相對較好4),但視功能恢復可能有限3)。早發型即使進行適當治療,許多患者在青少年時期也會達到法定盲。
各器官的附加治療
Section titled “各器官的附加治療”- 心臟:使用β受體阻滯劑等治療心衰竭。重症病例可考慮LVAD(左心室輔助裝置)或心臟移植1)。
- 腎臟:使用ACE抑制劑或ARB進行腎臟保護2)。
Q
治療能否改善眼部症狀?
A
早發型即使代謝指標改善,黃斑病變和視網膜變性的視功能預後也常不良,有報導稱許多患者在青少年時期達到法定盲。晚發型對神經症狀的治療反應相對較好,但殘留的視功能障礙僅能有限恢復3)。早期診斷和早期治療可能帶來最佳預後。
6. 病理生理學與詳細發病機制
Section titled “6. 病理生理學與詳細發病機制”MMACHC蛋白在溶酶體中與鈷胺素-結合球蛋白(R)複合物結合,從鈷胺素上切下R基團,生成可在細胞內利用的通用鈷胺素中間體。該中間體隨後分支為兩條途徑。
- 粒線體途徑:合成AdoCbl,作為甲基丙二醯輔酶A變位酶的輔酶。
- 細胞質途徑:合成MeCbl,作為甲硫胺酸合成酶的輔酶。
當MMACHC功能喪失導致兩條途徑均受損時:
- MMA蓄積:AdoCbl缺乏→甲基丙二醯輔酶A變位酶活性降低→MMA蓄積→神經毒性(代謝性中風)
- Hcy蓄積:MeCbl缺乏→甲硫胺酸合成酶活性降低→Hcy蓄積→血管損傷、血栓形成2)、腎TMA2)
- 甲硫胺酸降低:Hcy轉化為甲硫胺酸障礙→S-腺苷甲硫胺酸(SAM)減少→甲基化障礙→視神經萎縮
對眼組織的影響方面,甲硫胺酸降低導致穀胱甘肽(GSH)合成障礙,降低視網膜色素上皮(RPE)的氧化壓力防禦能力,被認為是黃斑部病變的一個原因。高Hcy引起的血管內皮損傷損害視網膜和脈絡膜循環的機制也被推測。
7. 最新研究與未來展望(研究階段報告)
Section titled “7. 最新研究與未來展望(研究階段報告)”合併嚴重心衰竭的cblC患者的LVAD和心臟移植
Section titled “合併嚴重心衰竭的cblC患者的LVAD和心臟移植”Hjalmarssон等人(2024)報告了全球首例在合併心肌病的cblC患者中植入LVAD(左心室輔助裝置)並隨後進行心臟移植的病例1)。結合高劑量OH-Cbl(30 mg/日靜脈注射)、甜菜鹼、亞葉酸和左卡尼汀的代謝治療,代謝指標改善(MMA 97→8.1,Hcy 91→31.8),並獲得了良好結果。
研究表明,即使在心臟移植後,繼續對cblC進行代謝治療也是必不可少的1)。
新生兒篩檢的局限性與晚發型診斷的改進
Section titled “新生兒篩檢的局限性與晚發型診斷的改進”已有研究表明,部分晚發型和輕症病例在新生兒篩檢中被漏診4),強調在出現神經或精神症狀的年輕成人中,即使B12正常,也應測量Hcy和MMA的重要性3, 4)。
epi-cblC的發現
Section titled “epi-cblC的發現”已有報告因PRDX1基因突變導致MMACHC表觀遺傳沉默引起的「epi-cblC」。即使沒有基因突變也可能呈現cblC樣病態,因此需要重新考慮診斷演算法。
家族篩檢的重要性
Section titled “家族篩檢的重要性”Ailliet等人(2022)報告了透過遲發型cblC的家族篩檢早期發現無症狀純合子的經驗,強調了以先證者診斷為契機的積極家族篩檢的意義3)。
8. 參考文獻
Section titled “8. 參考文獻”- Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
- Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
- Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
- Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.