A deficiência de cobalamina C (tipo cblC) é um erro inato do metabolismo intracelular da vitamina B12 (cobalamina). O nome oficial é acidúria metilmalônica e homocistinúria tipo cblC (OMIM #277400). É o tipo mais comum, representando cerca de 80% de todos os distúrbios do metabolismo da cbl.
O padrão de herança é autossômico recessivo. A causa são mutações no gene MMACHC (1p34.2). A proteína MMACHC é responsável pelos estágios iniciais do metabolismo intracelular da cobalamina, e sua perda de função prejudica a síntese tanto de adenosilcobalamina (AdoCbl) quanto de metilcobalamina (MeCbl).
A incidência estimada é de 1:100.000 a 1:200.000 3, 4), mas um estudo na província de Shandong, China, relatou uma alta frequência de 1:3.920. Nos Estados Unidos, a triagem neonatal (NBS) tornou-se alvo desde o início dos anos 2000 4).
De acordo com a idade de início, é dividido em tipo de início precoce (no primeiro ano de vida, 86-88% do total) e tipo de início tardio (após 1 ano de idade). Mais de 160 casos de início tardio foram relatados, e mais de 30 casos de início na idade adulta (acima de 18 anos) 3).
QA deficiência de cobalamina C pode ser detectada pela triagem neonatal?
A
A detecção precoce é possível através do NBS usando a elevação da propionilcarnitina (C3) no sangue como indicador. No entanto, casos de início tardio ou leves podem escapar do NBS 4), e se houver suspeita clínica apesar do NBS normal, são necessários exames adicionais de Hcy plasmático e MMA urinário.
Os achados de fundo de olho diferem amplamente entre os tipos de início precoce e tardio.
Tipo de Início Precoce
Maculopatia: O achado mais característico. Pode aparecer a partir do 35º dia de vida.
Maculopatia em olho de boi: Padrão característico de atrofia coriorretiniana central com anel de pigmento.
Alterações semelhantes à retinite pigmentosa: Ocorre degeneração retiniana extensa.
Atrofia do nervo óptico: Presente em casos avançados.
Angiografia fluoresceínica do fundo (FAF): Padrão de hipoautofluorescência foveal e hiperautofluorescência nas bordas.
Tipo tardio
Fundo de olho frequentemente normal: Muitas vezes não apresenta achados oculares graves3, 4).
Achados de RM da medula espinhal: Lesões hiperintensas no funículo posterior são características4).
Lesões da substância branca na RM cerebral: Observadas em alguns casos do tipo tardio3).
Retinopatia hipertensiva: Pode ocorrer em casos graves com complicações renais (microangiopatia trombótica)2).
As complicações sistêmicas incluem acidose metabólica, hipotonia, microcefalia, cardiomiopatia (50%)1) e microangiopatia trombótica renal (TMA)2).
QO tipo tardio também apresenta complicações oculares?
A
No tipo tardio, maculopatia grave e degeneração retiniana como no tipo precoce são raras, e o fundo de olho é frequentemente normal3, 4). No entanto, em casos graves com complicações renais, pode ocorrer retinopatia hipertensiva2). A melhora dos sintomas neurológicos geralmente é boa com o tratamento, mas foram relatados casos com déficit visual residual3).
A deficiência de cobalamina C é uma doença autossômica recessiva causada por mutações no gene MMACHC. Mais de 80 mutações foram relatadas, e o fenótipo varia amplamente dependendo do tipo de mutação.
A frequência das principais mutações e os fenótipos correspondentes são mostrados abaixo.
Mutação
Frequência/Linhagem
Fenótipo Principal
c.271dupA
40-61% · Europeus
Início precoce · Mais grave
c.394C>T
Cerca de 20%
Tipo tardio
c.609G>A
48-55% · Asiáticos orientais
Lactentes a crianças
Outras mutações principais:
c.331C>T: 5-9% em franco-canadenses1).
c.566G>A: Associado ao tipo tardio3).
c.484G>T: Associado a doença multiorgânica em lactentes2).
c.271dupA + c.449T>A (heterozigoto composto): Foram relatados casos apresentando tipo tardio4).
Existe também o “epi-cblC” causado pelo silenciamento epigenético do MMACHC devido a mutação no gene PRDX1, levando a uma condição secundária semelhante a cblC.
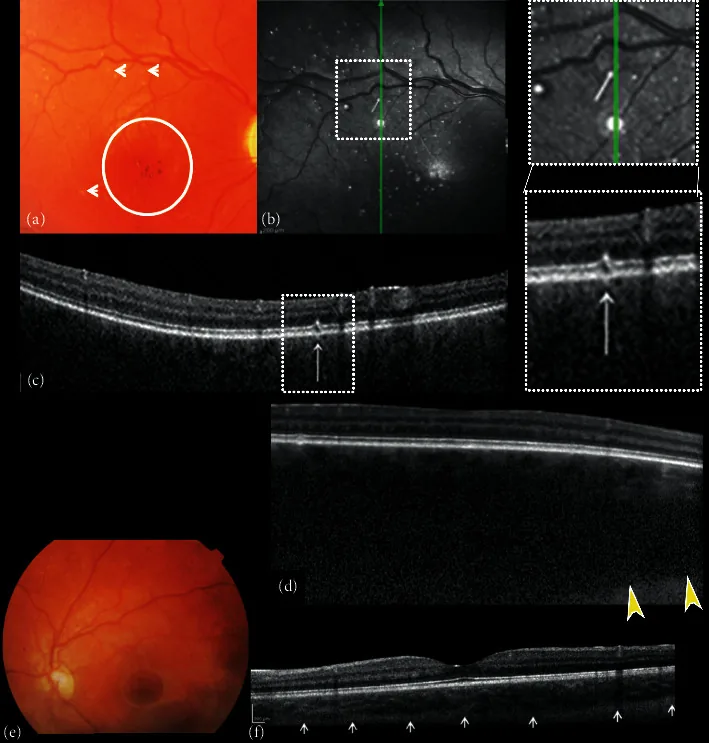
Composição de fotografias coloridas do fundo e tomografia de coerência óptica de domínio espectral (SDOCT) dos olhos direito e esquerdo. (a) A imagem do fundo direito mostra escavação e palidez do disco óptico, ausência de tesselação, hemangioma coroidal difuso, hiper-hipopigmentação da área foveal com reflexo foveal ausente (círculo) e pequenas alterações brancas em forma de ponto semelhantes a “microdrusas” (setas). (b) A reflectância de infravermelho próximo (NIR) do olho direito mostra múltiplos pontos hiper-refletivos circundados por um anelo hiporrefletivo correspondente às alterações de ponto branco semelhantes a “microdrusas” do polo posterior observadas na oftalmoscopia. A varredura B-scan SDOCT (c) sobre os pontos hiper-refletivos mostra alterações focais da camada do epitélio pigmentar da retina (RPE)-fotorreceptor.
Triagem Neonatal (NBS): Elevação da C3 propionilcarnitina por espectrometria de massa em tandem como indicador. No entanto, casos leves e de início tardio podem ser perdidos 4).
Os achados laboratoriais que indicam anormalidade metabólica são centrais para o diagnóstico.
Item do Exame
Achados Típicos
Significado
Hcy Plasmática
Muito elevado
Triagem
MMA urinário
Elevado
Confirmação de bloqueio metabólico
Teste genético
Mutação MMACHC
Diagnóstico definitivo
Valores numéricos relatados: Hcy plasmático 101,5–1700,5 µmol/L2, 3), MMA urinário 85–802 mmol/mol3, 4). A vitamina B12 sérica é frequentemente normal na forma de início tardio (448–625 pg/mL)3, 4).
É importante diferenciar entre deficiência dietética de B12, anemia perniciosa, outros distúrbios do metabolismo da cbl (como cblD, cblF) e deficiência de MTHFR3, 4). A combinação de Hcy elevado e MMA elevado é característica de cblC, enquanto apenas Hcy elevado (MMA normal) sugere deficiência de MTHFR, etc.
QÉ possível ter cblC mesmo com vitamina B12 sérica normal?
A
Sim. Na cblC de início tardio, a B12 sérica frequentemente está na faixa normal3, 4), e não deve ser descartada apenas com B12 normal. Em pacientes jovens com sintomas neurológicos ou psiquiátricos, Hcy e MMA plasmáticos devem ser medidos e, se elevados, a análise genética do MMACHC é necessária.
O tratamento principal é a administração parenteral de hidroxicobalamina (OH-Cbl), combinada com betaína, ácido folínico e L-carnitina em terapia multidrogas.
A administração parenteral é a base. Dose aproximada: 1 mg/dia ou 0,3 mg/kg/dia por via intramuscular (IM)4). Doses relatadas:
Em um caso adulto de início tardio, Ailliet et al. (2022) administraram 25 mg/dia por via subcutânea (SC) e obtiveram melhora dos sintomas neurológicos3). No caso de Goyne et al. (2023), iniciou-se com 5 mg/dia IM4). No caso de Hjalmarsson et al. (2024) com insuficiência cardíaca grave, foi administrado 30 mg/dia por via intravenosa (IV)1).
Promove a remetilação da homocisteína em metionina. Dose aproximada: 250 mg/kg/dia divididos em 3 doses4). Doses relatadas: 100 mg/kg/dia2), 1 g × 3 vezes4), 2-3 g × 3 vezes1).
O objetivo do tratamento é reduzir MMA e Hcy e normalizar os indicadores metabólicos. O tratamento adequado resulta em redução acentuada de Hcy:
Em um caso grave com TMA renal de Akar et al. (2024), Hcy normalizou de 1.700,5 para 4,6 µmol/L após início do tratamento2). Em um caso com cardiomiopatia de Hjalmarsson et al. (2024), MMA melhorou de 97 para 8,1 mmol/mol e Hcy de 91 para 31,8 µmol/L1).
No tipo de início tardio, a resposta ao tratamento é relativamente boa4), mas a recuperação da função visual pode ser limitada3). No tipo de início precoce, mesmo com tratamento adequado, muitos pacientes atingem cegueira legal na adolescência.
Coração: Tratamento de insuficiência cardíaca com betabloqueadores, etc. Em casos graves, LVAD (dispositivo de assistência ventricular esquerda) ou transplante cardíaco podem ser considerados1).
Rins: Proteção renal com inibidores da ECA ou BRA2).
QOs sintomas oculares melhoram com o tratamento?
A
No tipo de início precoce, mesmo com melhora dos indicadores metabólicos, o prognóstico da função visual para maculopatia e degeneração retiniana é frequentemente ruim, e casos de cegueira legal na adolescência foram relatados. No tipo de início tardio, a resposta dos sintomas neurológicos ao tratamento é relativamente boa, mas o comprometimento visual residual melhora apenas de forma limitada3). O diagnóstico e tratamento precoces podem proporcionar o melhor prognóstico.
A proteína MMACHC se liga ao complexo cobalamina-haptocorina (R) no lisossomo, separando o grupo R da cobalamina para produzir um intermediário comum de cobalamina utilizável intracelularmente. Esse intermediário então se ramifica em duas vias.
Via mitocondrial: Sintetiza AdoCbl e funciona como coenzima para a metilmalonil CoA mutase.
Via citoplasmática: Sintetiza MeCbl e atua como coenzima da metionina sintase.
Quando ambas as vias são prejudicadas pela perda de função do MMACHC:
Acúmulo de MMA: Deficiência de AdoCbl → redução da atividade da metilmalonil-CoA mutase → acúmulo de MMA → neurotoxicidade (acidente vascular metabólico)
Acúmulo de Hcy: Deficiência de MeCbl → redução da atividade da metionina sintase → acúmulo de Hcy → dano vascular e trombose2) e TMA renal2)
Redução de metionina: Conversão prejudicada de Hcy em metionina → redução de S-adenosilmetionina (SAM) → metilação prejudicada → atrofia do nervo óptico
Quanto aos efeitos nos tecidos oculares, acredita-se que a síntese prejudicada de glutationa (GSH) devido à redução de metionina diminui a defesa antioxidante do epitélio pigmentar da retina (EPR), contribuindo para a degeneração macular. Também se especula que o dano endotelial vascular causado pelo Hcy elevado prejudica a circulação retiniana e coroidal.
7. Pesquisas recentes e perspectivas futuras (relatos em fase de pesquisa)
Hjalmarssон et al. (2024) relataram o primeiro caso mundial de implante de LVAD (dispositivo de assistência ventricular esquerda) seguido de transplante cardíaco em paciente cblC com cardiomiopatia1). Combinado com terapia metabólica com OH-Cbl em alta dose (30 mg/dia IV), betaína, ácido folínico e L-carnitina, resultou em melhora dos indicadores metabólicos (MMA 97→8,1, Hcy 91→31,8) e bom desfecho.
O caso demonstrou que a continuação da terapia metabólica para cblC após o transplante cardíaco é essencial1).
Limitações da triagem neonatal e melhora do diagnóstico da forma tardia
Estudos mostram que alguns casos de forma tardia ou leve podem ser perdidos na triagem neonatal4), enfatizando a importância de medir Hcy e MMA em adultos jovens com sintomas neurológicos ou psiquiátricos, mesmo com B12 normal3, 4).
Foi relatado o “epi-cblC” causado pelo silenciamento epigenético do MMACHC devido a mutação no gene PRDX1. Como pode apresentar fenótipos semelhantes ao cblC mesmo sem mutação genética, é necessária uma reconsideração do algoritmo diagnóstico.
Ailliet et al. (2022) relataram a experiência de detecção precoce de homozigotos assintomáticos por meio de triagem familiar em cblC de início tardio, e enfatizaram a importância da triagem familiar ativa após o diagnóstico do probando 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.