Der Cobalamin-C-Mangel (cblC-Typ) ist eine angeborene Stoffwechselstörung des intrazellulären Vitamin-B12 (Cobalamin)-Stoffwechsels. Die vollständige Bezeichnung lautet Methylmalonazidurie und Homocystinurie, cblC-Typ (OMIM #277400). Es ist der häufigste Typ unter den cbl-Stoffwechselstörungen und macht etwa 80 % aller Fälle aus.
Der Erbgang ist autosomal-rezessiv. Ursache sind Mutationen im MMACHC-Gen (1p34.2). Das MMACHC-Protein ist an den frühen Schritten des intrazellulären Cobalamin-Stoffwechsels beteiligt; sein Funktionsverlust führt zu einer gestörten Synthese sowohl von Adenosylcobalamin (AdoCbl) als auch von Methylcobalamin (MeCbl).
Die geschätzte Inzidenz liegt bei 1:100.000 bis 1:200.000 3, 4), aber eine Studie in der chinesischen Provinz Shandong berichtete eine hohe Häufigkeit von 1:3.920. In den USA ist die Erkrankung seit Anfang der 2000er Jahre Gegenstand des Neugeborenen-Screenings (NBS) 4).
Nach dem Erkrankungsalter wird zwischen der früh beginnenden Form (innerhalb des ersten Lebensjahres, 86–88 % aller Fälle) und der späten Form (nach dem 1. Lebensjahr) unterschieden. Es wurden über 160 Fälle der späten Form und über 30 Fälle mit Erwachsenenbeginn (≥18 Jahre) berichtet 3).
QKann ein Cobalamin-C-Mangel durch das Neugeborenen-Screening entdeckt werden?
A
Ein NBS basierend auf erhöhtem C3-Propionylcarnitin im Blut ermöglicht eine Früherkennung. Allerdings können späte oder milde Formen dem NBS entgehen 4); bei klinischem Verdacht trotz normalem Screening sind zusätzliche Tests (Plasma-Hcy, Urin-MMA) erforderlich.
Das klinische Bild variiert stark je nach Erkrankungsalter.
Augensymptome der frühen Form:
Nystagmus: Das früheste Augenmerkmal. Tritt bei etwa 70–76 % der Patienten auf.
Sehverschlechterung: Schreitet seit dem Säuglingsalter fort und führt selbst mit angemessener Behandlung oft im Teenageralter zur gesetzlichen Blindheit.
Schielen (Strabismus): Tritt häufig zusammen mit Nystagmus auf.
Hauptsymptome der späten Form (extraokulär):
Schwäche und Taubheit der Beine: Aufgrund einer Schädigung der Hinterstränge des Rückenmarks4).
Gangstörungen und psychische Symptome: Bei Erwachsenen mit spätem Beginn berichtet3, 4).
QTreten auch bei der Spätmanifestation Augenkomplikationen auf?
A
Bei der Spätmanifestation sind schwere Makulopathie und Netzhautdegeneration wie bei der frühen Form selten, der Fundus ist oft normal3, 4). Bei schweren Fällen mit Nierenkomplikationen kann jedoch eine hypertensive Retinopathie auftreten2). Die neurologischen Symptome sprechen meist gut auf die Behandlung an, es wurden jedoch auch Fälle mit verbleibender Sehbeeinträchtigung berichtet3).
Cobalamin-C-Mangel ist eine autosomal-rezessive Erkrankung, die durch Mutationen im MMACHC-Gen verursacht wird. Es wurden über 80 Mutationen beschrieben, und der Phänotyp variiert stark je nach Mutationstyp.
Die Häufigkeit der wichtigsten Mutationen und die entsprechenden Phänotypen sind unten aufgeführt.
Mutation
Häufigkeit / Abstammung
Hauptphänotyp
c.271dupA
40–61 % bei Europäern
Früher Beginn, schwerste Form
c.394C>T
Etwa 20 %
Spätmanifestation
c.609G>A
48–55 % bei Ostasiaten
Säuglings- bis Kindesalter
Weitere Hauptmutationen:
c.331C>T: 5–9 % bei Frankokanadiern1).
c.566G>A: mit Spätmanifestation assoziiert3).
c.484G>T: mit Multiorganerkrankung im Säuglingsalter assoziiert2).
c.271dupA + c.449T>A (compound heterozygot): Fälle mit Spätmanifestation wurden berichtet4).
Es existiert auch ein „epi-cblC“, verursacht durch epigenetisches Silencing von MMACHC aufgrund einer PRDX1-Genmutation, das ein sekundäres cblC-ähnliches Krankheitsbild hervorruft.
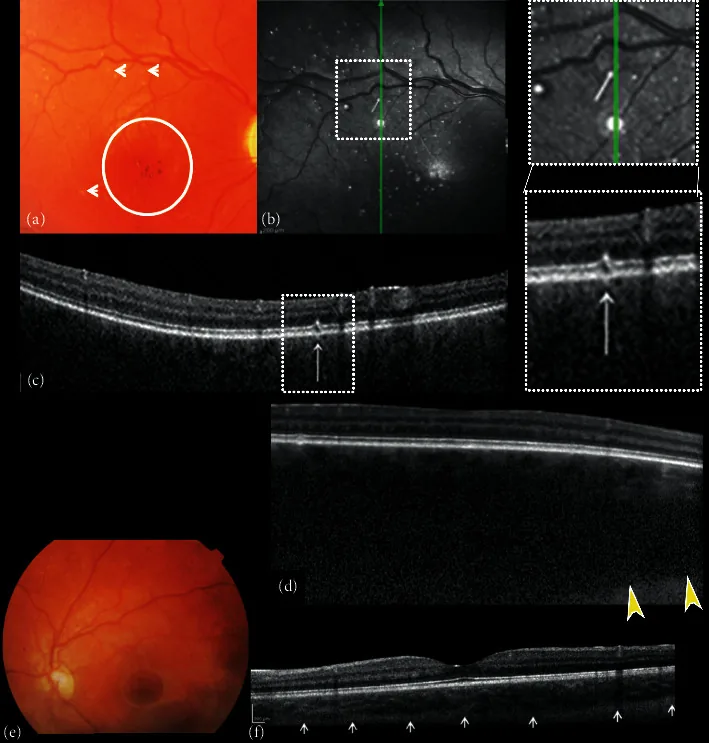
Zusammensetzung aus Fundus-Farbfotografien und spektraldomänenoptischer Kohärenztomographie (SDOCT) des rechten und linken Auges. (a) Das rechte Fundusbild zeigt eine Exkavation und Blässe der Papille, Fehlen von Tessellation, diffuses Aderhaut-Hämangiom, Hypo-Hyperpigmentierung der Fovea mit fehlendem Fovealreflex (Kreis) und kleine weiße punktförmige „Mikrodrusen-ähnliche“ Veränderungen (Pfeile). (b) Nahinfrarot-Reflexion (NIR) des rechten Auges zeigt mehrere hyperreflektive Punkte, umgeben von einem hyporeflektiven Ring, die den bei der Ophthalmoskopie beobachteten kleinen weißen punktförmigen „Mikrodrusen-ähnlichen“ Veränderungen des hinteren Pols entsprechen. Der B-Scan-Querschnitts-SDOCT (c) über den hyperreflektiven Punkten zeigt fokale Veränderungen der retinalen Pigmentepithel (RPE)-Photorezeptorschicht.
Neugeborenen-Screening (NBS) : Erhöhtes C3-Propionylcarnitin mittels Tandem-Massenspektrometrie ist ein Indikator. Leichte und spät beginnende Formen können jedoch übersehen werden 4).
Laborbefunde, die auf Stoffwechselstörungen hinweisen, stehen im Mittelpunkt der Diagnose.
Testparameter
Typischer Befund
Bedeutung
Plasma-Hcy
Deutlich erhöht
Screening
Urin-MMA
Erhöht
Bestätigung des metabolischen Blocks
Gentest
MMACHC-Mutation
Bestätigungsdiagnose
Berichtete spezifische Werte: Plasma-Hcy 101,5–1700,5 µmol/L2, 3), Urin-MMA 85–802 mmol/mol3, 4). Serum-Vitamin B12 ist bei spätem Beginn oft im Normbereich (448–625 pg/mL)3, 4).
Die Abgrenzung zur alimentären B12-Mangelanämie, perniziösen Anämie, anderen cbl-Stoffwechselstörungen (cblD, cblF usw.) und zum MTHFR-Mangel ist wichtig3, 4). Die Kombination aus erhöhtem Hcy und erhöhtem MMA ist charakteristisch für cblC, während ein isoliert erhöhtes Hcy (MMA normal) auf einen MTHFR-Mangel hindeutet.
QIst ein cblC möglich, obwohl das Serum-Vitamin B12 normal ist?
A
Ja. Bei spätmanifestem cblC liegt das Serum-B12 oft im Normbereich3, 4); ein normales B12 allein schließt die Erkrankung nicht aus. Bei jungen Patienten mit neurologischen oder psychiatrischen Symptomen sollten Plasma-Hcy und MMA gemessen werden; sind sie erhöht, ist eine genetische Analyse des MMACHC-Gens erforderlich.
Die Hauptbehandlung ist die parenterale Gabe von Hydroxocobalamin (OH-Cbl), kombiniert mit Betain, Folinsäure und L-Carnitin in einer Mehrfachtherapie.
Die parenterale Gabe ist Standard. Dosierungsrichtwert: 1 mg/Tag oder 0,3 mg/kg/Tag intramuskulär (IM)4). Berichtete Dosierungen:
Ailliet et al. (2022) verabreichten bei einem spätmanifesten Erwachsenen 25 mg/Tag subkutan (SC) und erzielten eine Besserung der neurologischen Symptome3). Goyne et al. (2023) begannen mit 5 mg/Tag IM4). Hjalmarsson et al. (2024) gaben bei einem Patienten mit schwerer Herzinsuffizienz 30 mg/Tag intravenös (IV)1).
Fördert die Remethylierung von Homocystein zu Methionin. Dosierungsrichtwert: 250 mg/kg/Tag in 3 Dosen4). Berichtete Dosierungen: 100 mg/kg/Tag2), 1 g × 34), 2-3 g × 31).
Behandlungsziele sind die Senkung von MMA und Hcy sowie die Normalisierung der Stoffwechselparameter. Eine angemessene Behandlung führt zu einer deutlichen Senkung des Hcy:
Akar et al. (2024) berichten über einen schweren Fall mit renaler TMA, bei dem Hcy nach Behandlungsbeginn von 1.700,5 auf 4,6 µmol/L normalisiert wurde2). Hjalmarssон et al. (2024) beschreiben einen Fall mit Kardiomyopathie, bei dem MMA von 97 auf 8,1 mmol/mol und Hcy von 91 auf 31,8 µmol/L verbessert wurden1).
Bei spätmanifesten Formen ist das Ansprechen auf die Behandlung relativ gut4), die Erholung der Sehfunktion kann jedoch begrenzt sein3). Bei frühmanifesten Formen erreichen viele Patienten trotz angemessener Behandlung im Teenageralter die gesetzliche Blindheit.
Herz: Behandlung der Herzinsuffizienz mit Betablockern usw. In schweren Fällen kann ein LVAD (linksventrikuläres Unterstützungssystem) oder eine Herztransplantation in Betracht gezogen werden1).
Nieren: Nierenschutz durch ACE-Hemmer oder ARB2).
QBessern sich die Augensymptome durch die Behandlung?
A
Bei frühmanifesten Formen ist die Sehprognose für Makulopathie und Netzhautdegeneration trotz Verbesserung der Stoffwechselparameter oft schlecht, und es wird über Fälle von gesetzlicher Blindheit im Teenageralter berichtet. Bei spätmanifesten Formen ist das Ansprechen der neurologischen Symptome auf die Behandlung relativ gut, aber die verbleibende Sehbehinderung erholt sich nur begrenzt3). Frühe Diagnose und frühzeitiger Behandlungsbeginn könnten die beste Prognose ermöglichen.
Das MMACHC-Protein bindet im Lysosom an den Cobalamin-Haptocorin(R)-Komplex, spaltet die R-Gruppe vom Cobalamin ab und erzeugt ein gemeinsames Cobalamin-Zwischenprodukt, das in der Zelle verwendet werden kann. Dieses Zwischenprodukt verzweigt sich dann in zwei Wege.
Mitochondrialer Weg: Synthese von AdoCbl, das als Coenzym der Methylmalonyl-CoA-Mutase fungiert.
Zytosolischer Weg : Synthese von MeCbl, das als Coenzym der Methioninsynthase dient.
Wenn beide Wege durch einen Funktionsverlust von MMACHC gestört sind:
Hcy-Akkumulation : MeCbl-Mangel → verminderte Aktivität der Methioninsynthase → Hcy-Akkumulation → Gefäßschäden und Thrombose2)·renale TMA2)
Methioninabfall : Gestörte Umwandlung von Hcy zu Methionin → vermindertes S-Adenosylmethionin (SAM) → Methylierungsstörung → Optikusatrophie
Hinsichtlich der Augenwirkungen wird angenommen, dass eine verminderte Glutathion (GSH)-Synthese aufgrund niedrigen Methionins die oxidative Stressabwehr des retinalen Pigmentepithels (RPE) verringert und zur Makuladegeneration beiträgt. Es wird auch vermutet, dass eine durch hohes Hcy verursachte vaskuläre Endothelschädigung die retinale und choroidale Zirkulation beeinträchtigt.
7. Aktuelle Forschung und Zukunftsaussichten (Berichte aus der Forschungsphase)
Hjalmarssон et al. (2024) berichteten über den weltweit ersten Fall einer LVAD-Implantation (linksventrikuläres Unterstützungssystem) gefolgt von einer Herztransplantation bei einem cblC-Patienten mit Kardiomyopathie1). In Kombination mit einer metabolischen Therapie mit hochdosiertem OH-Cbl (30 mg/Tag i.v.), Betain, Folinsäure und L-Carnitin wurden eine Verbesserung der metabolischen Parameter (MMA 97→8,1, Hcy 91→31,8) und ein gutes Ergebnis erzielt.
Es wurde gezeigt, dass die Fortsetzung der metabolischen Therapie für cblC auch nach der Herztransplantation unerlässlich ist1).
Grenzen des Neugeborenen-Screenings und Verbesserung der Diagnose bei spätmanifesten Formen
Es wurde gezeigt, dass spätmanifeste und milde Formen im Neugeborenen-Screening übersehen werden können4), und die Bedeutung der Messung von Hcy und MMA bei jungen Erwachsenen mit neurologischen oder psychiatrischen Symptomen, selbst bei normalem B12, wird betont3, 4).
Der durch epigenetisches Silencing von MMACHC aufgrund einer PRDX1-Genmutation verursachte „epi-cblC“ wurde berichtet. Da auch ohne Genmutation ein cblC-ähnliches Krankheitsbild auftreten kann, ist eine Überarbeitung des Diagnosealgorithmus erforderlich.
Ailliet et al. (2022) berichteten über die Erfahrung der Früherkennung eines asymptomatischen Homozygoten durch Familienscreening bei spätmanifestem cblC und betonten die Bedeutung eines aktiven Familienscreenings nach der Diagnose des Probanden 3).
Clara Hjalmarsson, Charlotte Backelin, Anders Thoren, Niklas Bergh, Jennifer L. Sloan, Irini Manoli, Charles P. Venditti, Göran Dellgren. Severe heart failure in a unique case of cobalamin-C-deficiency resolved with LVAD implantation and subsequent heart transplantation. Molecular Genetics and Metabolism Reports. 2024;39:101089. doi:10.1016/j.ymgmr.2024.101089.
Akar HT, Yıldız H, Öztürk Z, Karakaya D, Sezer A, Olgaç A. Case presentation: a severe case of cobalamin c deficiency presenting with nephrotic syndrome, malignant hypertension and hemolytic anemia. BMC nephrology. 2024;25(1):217. doi:10.1186/s12882-024-03656-1. PMID:38977946; PMCID:PMC11232354.
Ailliet S, Vandenberghe R, Schiemsky T, et al. A case of vitamin B12 deficiency neurological syndrome in a young adult due to late-onset cobalamin C (CblC) deficiency: a diagnostic challenge. Biochem Med (Zagreb). 2022;32(2):020802. doi:10.11613/bm.2022.020802.
Christopher Goyne, Leena Kansal. Pearls & Oy-sters: Late-Onset Cobalamin C Deficiency Presenting With Subacute Combined Degeneration. Neurology. 2023;100(10):486-489. doi:10.1212/wnl.0000000000201695.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.