Alcaptonúria (AKU) é um distúrbio raro do metabolismo de aminoácidos, herdado de forma autossômica recessiva, causado pela deficiência da enzima homogentisato 1,2-dioxigenase, que catalisa a degradação do ácido homogentísico (HGA), um intermediário na via de degradação da tirosina. Essa deficiência leva ao acúmulo de HGA no corpo. O gene causador está localizado no cromossomo 3 (3q2).
Em 1902, Sir Archibald Garrod relatou a doença como uma condição hereditária e propôs pela primeira vez o conceito de “erros inatos do metabolismo”. A incidência global é estimada em 1 por 250.000 a 1.000.000 de pessoas 2). Na Eslováquia e na República Dominicana, a incidência chega a 1 por 19.000 pessoas. No Japão, a doença é extremamente rara.
A AKU é caracterizada pela tríade clássica: urina escura, ocronose e artropatia ocronótica. O HGA forma polímeros semelhantes à melanina através de um processo de oxidação mediado pelo ácido benziquinona acético, depositando-se no tecido conjuntivo, causando doença multissistêmica. A ocronose ocular geralmente aparece por volta dos 30 anos de idade e frequentemente é o sinal inicial da doença.
QO que é ocronose?
A
Ocronose é uma condição na qual polímeros semelhantes à melanina derivados do HGA se depositam no tecido conjuntivo, causando descoloração azul-escura. O nome deriva dessa coloração. Os depósitos ocorrem no tecido conjuntivo em todo o corpo, incluindo esclera, cartilagem auricular, cartilagem articular, válvulas cardíacas e rins. Nos olhos, a esclera, conjuntiva e córnea são afetadas. A ocronose exógena (devido a medicamentos tópicos como hidroquinona) difere da ocronose endógena (hereditária) na AKU.
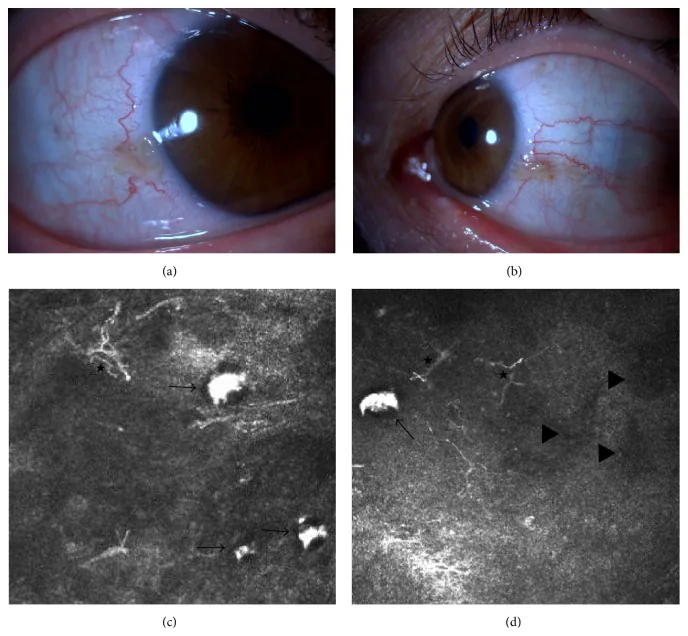
Elif Demirkilinc Biler, Suzan Guven Yilmaz, Melis Palamar, Pedram Hamrah, et al. In Vivo Confocal Microscopy and Anterior Segment Optic Coherence Tomography Findings in Ocular Ochronosis 2015 Dec 15 Case Rep Ophthalmol Med. 2015 Dec 15; 2015:592847 Figure 3. PMCID: PMC4693010. License: CC BY.
Fotografias clínicas da conjuntiva e imagens de microscopia confocal in vivo dos olhos direito e esquerdo no caso 2 de alcaptonúria. A imagem superior esquerda (a) mostra a conjuntiva nasal do olho direito, a imagem superior direita (b) mostra a conjuntiva nasal do olho esquerdo. As imagens inferiores (c) e (d) mostram acúmulo subconjuntival de ácido homogentísico (seta preta), células dendríticas (estrela) e áreas hiporrefletivas (triângulo preto).
A ocronose dos tecidos oculares geralmente aparece por volta dos 30 anos e frequentemente é um sinal precoce de AKU.
Achados Oculares Frequentes
Pigmentação azul-escura da esclera (sinal de Osler): Ocorre frequentemente próximo ao limbo corneano nos lados nasal e temporal da fenda palpebral. É particularmente observada no local de inserção do músculo reto lateral.
Depósitos corneanos em gota de óleo: Depósitos puntiformes ao nível da membrana de Bowman. Progressivos ao longo do tempo.
Pigmentação conjuntival: Pode estar associada a vasos conjuntivais dilatados.
Padrões de pigmentação escleral: Quatro padrões foram relatados: vermiforme, tipo pinguécula, pontilhado e laminar.
Achados Oculares Relativamente Raros
Astigmatismo corneano: Pode ocorrer astigmatismo progressivo devido ao afinamento periférico da córnea ao longo do eixo da lesão.
A pigmentação da esclera e conjuntiva por si só geralmente não afeta diretamente a acuidade visual. No entanto, se a deposição de pigmento na córnea progredir ou ocorrer glaucoma devido ao acúmulo de pigmento no ângulo da câmara anterior, pode levar à diminuição da visão. Recomenda-se acompanhamento com exames oftalmológicos regulares.
A AKU é causada por uma deficiência na atividade da enzima homogentisato 1,2-dioxigenase devido a uma mutação no gene HGD (3q2). Esta enzima é produzida principalmente nos hepatócitos e catalisa a degradação do HGA na via de degradação da tirosina. A deficiência enzimática leva ao acúmulo de HGA no corpo e à sua excreção em grandes quantidades na urina (1-8 g por dia) 1).
O HGA deposita-se nos tecidos colágenos, especialmente no nariz, orelhas, bochechas, conjuntiva, locais de inserção muscular, córnea e esclera.
Quantificação de HGA na urina: Padrão-ouro. Medido por cromatografia gasosa 1)
Teste de alcalinização da urina: Confirmar o escurecimento da cor da urina
Teste genético HGD: Determinar homozigoto ou heterozigoto composto
Biópsia da lesão ocular: Mostrar degeneração semelhante à elastose na coloração H&E
Se a cromatografia gasosa não estiver disponível, podem ser usados como substitutos o teste de Benedict, teste de hidróxido de sódio, teste de nitrato de prata ou teste de cloreto férrico 1).
Não houve terapia curativa para AKU por muito tempo, mas com o advento da nitisinona, a terapia modificadora da doença tornou-se possível.
Nitisinona: Inibe a 4-hidroxifenilpiruvato dioxigenase, reduzindo a produção de HGA. O estudo SONIA 2 (ensaio randomizado controlado multicêntrico internacional) mostrou que a dose de 10 mg/dia reduz a dor articular, melhora a densidade óssea e retarda a progressão da pigmentação ocular 2). A Agência Europeia de Medicamentos (EMA) aprovou seu uso para pacientes adultos com AKU em 2020 2)
Ácido ascórbico (Vitamina C): Tem fundamento teórico como antioxidante, mas a eficácia não foi comprovada 1)2)
Dieta pobre em proteínas: Restrição de ingestão de fenilalanina e tirosina. Difícil adesão a longo prazo em adultos 2)
Efeitos colaterais da nitisinona, como ceratopatia dendrítica devido à tirosinemia, foram relatados 1). Os efeitos colaterais podem ser prevenidos mantendo os níveis de tirosina abaixo de 500–600 μmol/L 1).
O estudo SONIA 2 mostrou que nitisinona 10 mg/dia reduziu a dor articular e vertebral, melhorou a densidade óssea (T-score) e inibiu a progressão da pigmentação ocular por 48 meses 2).
Exames oftalmológicos regulares: Para monitorar a progressão da pigmentação e o desenvolvimento de glaucoma
Correção refrativa: Considerada em caso de astigmatismo corneano progressivo
Manejo do glaucoma: Se ocorrer glaucoma devido ao acúmulo de pigmento no ângulo, realizar tratamento adequado
QO que é nitisinona?
A
Nitisinona é um medicamento que inibe a 4-hidroxifenilpiruvato dioxigenase, suprimindo a produção de HGA. Originalmente aprovado para tratar tirosinemia hereditária tipo 1. O estudo SONIA 1 mostrou que 8 mg/dia reduziu o HGA urinário de 24h em 98,8% 2). O estudo SONIA 2 confirmou a inibição da progressão dos sintomas articulares e oculares, e foi aprovado na Europa em 2020 para pacientes adultos com AKU 2).
A homogentisato 1,2-dioxigenase é produzida nos hepatócitos e converte HGA em maleilacetoacetato na via de degradação da tirosina. A deficiência dessa enzima leva ao acúmulo de HGA no corpo.
O HGA forma polímeros semelhantes à melanina (pigmento de escurecimento tecidual) através de um processo de oxidação mediado pelo ácido benziquinona acético. Esse pigmento adere ao tecido conjuntivo, causando descoloração azul-escura e dano tecidual.
O HGA exibe toxicidade direta sobre os condrócitos, levando à necrose da cartilagem e destruição articular acelerada 2). Processo semelhante ocorre nas válvulas cardíacas, com prevalência de estenose aórtica atingindo 22,2% dos pacientes com AKU 2). O N-telopeptídeo do colágeno (NTx) urinário está elevado, contribuindo para o aumento da reabsorção óssea e osteoporose 2).
A AKU não encurta a expectativa de vida por si só, mas afeta significativamente a qualidade de vida devido à artropatia, complicações cardiovasculares e cálculos renais 2). A introdução precoce de nitisinona pode prevenir o acúmulo de HGA e inibir a progressão da ocronose 1)2). No entanto, o efeito da nitisinona na artropatia ocronótica avançada é limitado, tornando o diagnóstico e tratamento precoces importantes 1).
A pigmentação ocular não é não progressiva, mas sim se expande ao longo do tempo. O prognóstico visual é relativamente bom, a menos que haja glaucoma associado.
Bhatti IA, Saqib M, Rehman IU, Amjed S, Hashim HT, Butt AA. Managing Alkaptonuria in Absence of Appropriate Medication: A Case Report and Review of Literature. Clinical medicine & research. 2024;22(2):107-111. doi:10.3121/cmr.2024.1867. PMID:39231619; PMCID:PMC11374495.
Roopnarinesingh RC, Donlon NE, Reynolds JV. Alkaptonuria: clinical manifestations and an updated approach to treatment of a rare disease. BMJ Case Rep. 2021;14:e244240.
Gupta PC, Balamurugan R, Ram J. Ocular and systemic manifestations of alkaptonuria. QJM. 2019;112(5):369. PMID: 30476261.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.