Alkaptonurie (AKU) ist eine seltene autosomal-rezessiv vererbte Aminosäurestoffwechselstörung, bei der Homogentisinsäure (HGA) aufgrund eines Mangels an Homogentisat-1,2-Dioxygenase, die den Abbau von HGA katalysiert, im Körper akkumuliert. Das verantwortliche Gen befindet sich auf Chromosom 3 (3q2).
1902 wurde die Erkrankung von Sir Archibald Garrod als Erbkrankheit beschrieben, womit er erstmals das Konzept der „angeborenen Stoffwechselstörungen“ (inborn errors of metabolism) prägte. Die weltweite Inzidenz wird auf 1 pro 250.000 bis 1 Million geschätzt 2). In der Slowakei und der Dominikanischen Republik ist sie mit etwa 1 pro 19.000 häufiger. In Japan ist die Erkrankung extrem selten.
AKU ist gekennzeichnet durch die klassische Trias aus dunklem Urin, Ochronose und Ochronose-Arthropathie. HGA bildet über einen Oxidationsprozess mit Benzochinonessigsäure melaninähnliche Polymere, die sich im Bindegewebe ablagern und eine Multisystemerkrankung verursachen. Die okuläre Ochronose tritt im Durchschnitt um das 30. Lebensjahr auf und ist oft ein frühes Anzeichen der Erkrankung.
QWas ist Ochronose?
A
Ochronose ist ein Zustand, bei dem sich HGA-abstammende melaninähnliche Polymere im Bindegewebe ablagern und eine blau-schwarze Verfärbung verursachen. Der Name leitet sich von dieser Farbe ab. Sie lagert sich im gesamten Bindegewebe ab, einschließlich Sklera, Ohrknorpel, Gelenkknorpel, Herzklappen und Nieren. Am Auge sind Sklera, Bindehaut und Hornhaut betroffen. Im Gegensatz zur exogenen Ochronose (Hautverfärbung durch topische Medikamente wie Hydrochinon) ist die Ochronose bei AKU endogen (hereditär).
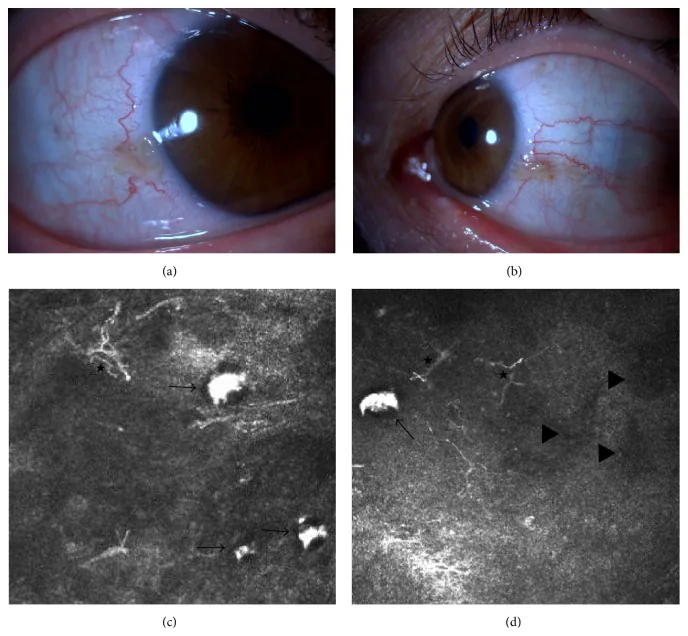
Elif Demirkilinc Biler, Suzan Guven Yilmaz, Melis Palamar, Pedram Hamrah, et al. In Vivo Confocal Microscopy and Anterior Segment Optic Coherence Tomography Findings in Ocular Ochronosis 2015 Dec 15 Case Rep Ophthalmol Med. 2015 Dec 15; 2015:592847 Figure 3. PMCID: PMC4693010. License: CC BY.
Klinische Fotos und In-vivo-Konfokalmikroskopie der Bindehaut des rechten und linken Auges bei Fall 2 der Alkaptonurie. Das obere linke Bild (a) zeigt die nasale Bindehaut des rechten Auges, das obere rechte Bild (b) die nasale Bindehaut des linken Auges. Die unteren Bilder (c) und (d) zeigen subkonjunktivale Homogentisinsäureablagerungen (schwarze Pfeile), dendritische Zellen (Sternchen) und hyporeflektive Bereiche (schwarze Dreiecke).
Die okuläre Melanose tritt in der Regel ab etwa 30 Jahren auf und ist oft ein frühes Zeichen der Alkaptonurie.
Häufige Augenmanifestationen
Blauschwarze Pigmentierung der Sklera (Osler-Zeichen): Tritt bevorzugt in der Lidspalte nasal und temporal nahe dem Limbus corneae auf. Besonders häufig an der Ansatzstelle des Musculus rectus lateralis.
Öltröpfchenartige Hornhautablagerungen: Fleckige Ablagerungen auf Höhe der Bowman-Membran. Im Verlauf fortschreitend.
Pigmentierung der Bindehaut: Kann von erweiterten Bindehautgefäßen begleitet sein.
Sklerale Pigmentierungsmuster: Es werden vier Typen beschrieben: wurmartig, pingueculaartig, punktförmig und lamellär.
Relativ seltene Augenmanifestationen
Hornhautastigmatismus: Durch Ausdünnung der peripheren Hornhaut im Bereich der Läsion kann ein fortschreitender Astigmatismus entstehen.
QBeeinträchtigen Augenpigmentierungen die Sehkraft?
A
Pigmentierungen der Sklera und Bindehaut selbst beeinträchtigen in der Regel nicht direkt die Sehkraft. Bei fortschreitender Ablagerung in der Hornhaut oder bei Entwicklung eines Glaukoms durch Pigmentansammlung im Kammerwinkel kann es jedoch zu einer Sehverschlechterung kommen. Regelmäßige augenärztliche Kontrollen werden empfohlen.
AKU wird durch einen Aktivitätsmangel der Homogentisat-1,2-Dioxygenase aufgrund einer Mutation im HGD-Gen (3q2) verursacht. Dieses Enzym wird hauptsächlich in Leberzellen produziert und katalysiert den Abbau von HGA im Tyrosinabbauweg. Der Enzymmangel führt zur Anhäufung von HGA im Körper und zur massiven Ausscheidung im Urin (1–8 g pro Tag) 1).
HGA lagert sich in Kollagengewebe ab, insbesondere in Nase, Ohren, Wangen, Bindehaut, Muskelansätzen, Hornhaut und Sklera.
HGA-Messung im Urin: Goldstandard. Messung mittels Gaschromatographie1)
Alkalisierungstest des Urins: Überprüfung der Verdunkelung der Urinfarbe
HGD-Gentest: Bestimmung von Homozygotie oder compound-Heterozygotie
Biopsie der Augenläsion: H&E-Färbung zeigt elastoseähnliche Degeneration
Wenn Gaschromatographie nicht verfügbar ist, können der Benedict-Test, Natriumhydroxid-Test, Silbernitrat-Test oder Eisen(III)-chlorid-Test als Ersatz verwendet werden1).
Eine kurative Behandlung für AKU war lange Zeit nicht verfügbar, aber mit dem Aufkommen von Nitisinon wurde eine krankheitsmodifizierende Therapie möglich.
Nitisinon: Hemmt die 4-Hydroxyphenylpyruvat-Dioxygenase und reduziert die HGA-Produktion. Die SONIA-2-Studie (internationale multizentrische randomisierte kontrollierte Studie) zeigte, dass eine Dosis von 10 mg/Tag Gelenkschmerzen reduziert, die Knochendichte verbessert und das Fortschreiten der Augenpigmentierung verlangsamt 2). Im Jahr 2020 wurde es von der Europäischen Arzneimittel-Agentur (EMA) für erwachsene AKU-Patienten zugelassen 2)
Ascorbinsäure (Vitamin C): Theoretische Begründung durch antioxidative Wirkung, aber Wirksamkeit nicht belegt 1)2)
Eiweißarme Ernährung: Einschränkung der Aufnahme von Phenylalanin und Tyrosin. Langfristige Einhaltung bei Erwachsenen schwierig 2)
Als Nebenwirkung von Nitisinon wurde eine dendritische Keratopathie aufgrund von Tyrosinämie berichtet 1). Es wird angenommen, dass die Aufrechterhaltung des Tyrosinspiegels unter 500–600 μmol/L diese Nebenwirkung verhindern kann 1).
In der SONIA-2-Studie zeigte Nitisinon 10 mg/Tag über 48 Monate eine Verringerung von Gelenk- und Wirbelsäulenschmerzen, eine Verbesserung der Knochendichte (T-Score) und eine Verlangsamung der Augenpigmentierung 2).
Regelmäßige augenärztliche Untersuchungen: Überwachung des Fortschreitens der Pigmentierung und der Entwicklung eines Glaukoms
Refraktionskorrektur: Wird bei fortschreitendem Hornhautastigmatismus in Betracht gezogen
Glaukommanagement: Bei Auftreten eines Glaukoms infolge der Pigmentablagerung im Kammerwinkel wird eine angemessene Behandlung durchgeführt.
QWas für ein Medikament ist Nitisinon?
A
Nitisinon hemmt die 4-Hydroxyphenylpyruvat-Dioxygenase und unterdrückt die Produktion von HGA. Ursprünglich wurde es zur Behandlung der hereditären Tyrosinämie Typ 1 zugelassen. Die SONIA-1-Studie zeigte, dass eine Dosis von 8 mg/Tag die 24-Stunden-HGA-Ausscheidung im Urin um 98,8 % senkte2). Die SONIA-2-Studie bestätigte die Verlangsamung des Fortschreitens von Gelenk- und Augensymptomen, und 2020 wurde es in Europa für erwachsene AKU-Patienten zugelassen2).
Die Homogentisat-1,2-Dioxygenase wird in Leberzellen produziert und wandelt HGA im Tyrosinabbauweg in Maleylacetoacetat um. Bei einem Defekt dieses Enzyms akkumuliert HGA im Körper.
HGA bildet über einen Oxidationsprozess mit Benzochinonessigsäure melaninähnliche Polymere (Gewebeschwärzungspigmente). Diese Pigmente haften an Bindegewebe und verursachen blau-schwarze Verfärbungen und Gewebeschäden.
HGA wirkt direkt toxisch auf Knorpelzellen, was zu Knorpelnekrose und beschleunigter Gelenkzerstörung führt2). Ein ähnlicher Prozess tritt an den Herzklappen auf, und die Prävalenz der Aortenklappenstenose erreicht 22,2 % bei AKU-Patienten2). Die Konzentration von kollagenem N-Telopeptid (NTx) im Urin ist erhöht, was zu gesteigerter Knochenresorption und Osteoporose beiträgt2).
AKU verkürzt die Lebenserwartung nicht direkt, beeinträchtigt jedoch die Lebensqualität erheblich durch Gelenkerkrankungen, kardiovaskuläre Komplikationen und Nierensteine 2). Die frühzeitige Einführung von Nitisinon kann die HGA-Akkumulation verhindern und das Fortschreiten der Ochronose möglicherweise aufhalten 1)2). Allerdings ist die Wirkung von Nitisinon bei bereits fortgeschrittener Ochronose-Arthropathie begrenzt, sodass eine frühzeitige Diagnose und Behandlung entscheidend sind 1).
Die Augenpigmentierung ist nicht nicht-progressiv, sondern nimmt im Laufe der Zeit zu. Sofern kein Glaukom auftritt, ist die Sehprognose relativ gut.
Bhatti IA, Saqib M, Rehman IU, Amjed S, Hashim HT, Butt AA. Managing Alkaptonuria in Absence of Appropriate Medication: A Case Report and Review of Literature. Clinical medicine & research. 2024;22(2):107-111. doi:10.3121/cmr.2024.1867. PMID:39231619; PMCID:PMC11374495.
Roopnarinesingh RC, Donlon NE, Reynolds JV. Alkaptonuria: clinical manifestations and an updated approach to treatment of a rare disease. BMJ Case Rep. 2021;14:e244240.
Gupta PC, Balamurugan R, Ram J. Ocular and systemic manifestations of alkaptonuria. QJM. 2019;112(5):369. PMID: 30476261.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.