L’alcaptonurie (AKU) est une maladie rare du métabolisme des acides aminés, à transmission autosomique récessive, causée par un déficit en homogentisate 1,2-dioxygénase, enzyme qui catalyse la dégradation de l’acide homogentisique (HGA), un intermédiaire de la voie de dégradation de la tyrosine, entraînant une accumulation d’HGA dans l’organisme. Le gène responsable est situé sur le chromosome 3 (3q2).
En 1902, Sir Archibald Garrod a décrit la maladie comme une maladie héréditaire et a proposé pour la première fois le concept d’« erreur innée du métabolisme ». L’incidence mondiale est estimée à 1 pour 250 000 à 1 000 000 de personnes 2). En Slovaquie et en République dominicaine, elle est plus fréquente, touchant environ 1 personne sur 19 000. Au Japon, la maladie est extrêmement rare.
L’AKU se caractérise classiquement par une triade : urines foncées, ochronose et arthropathie ochronotique. L’HGA forme des polymères de type mélanique via un processus d’oxydation impliquant l’acide benzoquinone acétique, qui se déposent dans les tissus conjonctifs, entraînant une maladie multisystémique. L’ochronose oculaire apparaît en moyenne vers l’âge de 30 ans et constitue souvent un signe précoce de la maladie.
QQu'est-ce que l'ochronose ?
A
L’ochronose est une condition où des polymères de type mélanique dérivés de l’HGA se déposent dans les tissus conjonctifs, provoquant une décoloration bleu-noir. Son nom provient de cette teinte. Les dépôts touchent les tissus conjonctifs de tout le corps, notamment la sclère, le cartilage auriculaire, le cartilage articulaire, les valves cardiaques et les reins. Au niveau oculaire, la sclère, la conjonctive et la cornée sont affectées. Contrairement à l’ochronose exogène (décoloration cutanée due à des médicaments topiques comme l’hydroquinone), l’ochronose de l’AKU est endogène (héréditaire).
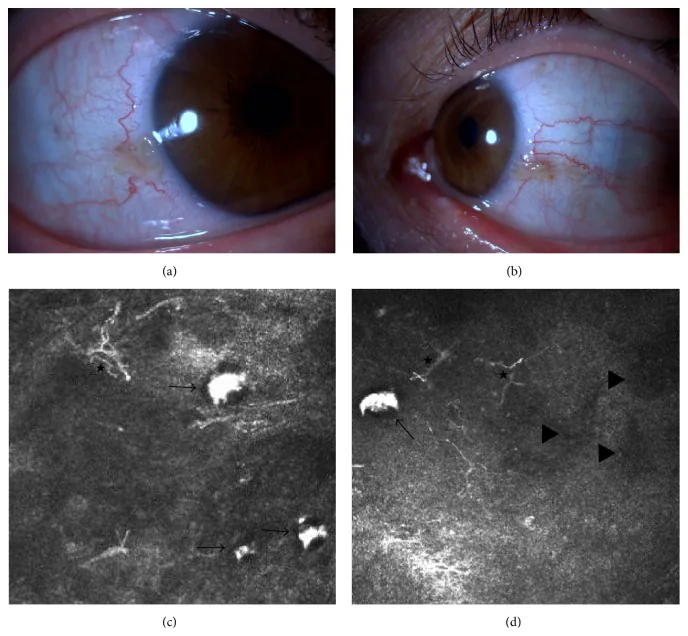
Elif Demirkilinc Biler, Suzan Guven Yilmaz, Melis Palamar, Pedram Hamrah, et al. In Vivo Confocal Microscopy and Anterior Segment Optic Coherence Tomography Findings in Ocular Ochronosis 2015 Dec 15 Case Rep Ophthalmol Med. 2015 Dec 15; 2015:592847 Figure 3. PMCID: PMC4693010. License: CC BY.
Photographies cliniques et images de microscopie confocale in vivo de la conjonctive de l’œil droit et de l’œil gauche chez un patient atteint d’alcaptonurie (cas 2). L’image en haut à gauche (a) montre la conjonctive nasale de l’œil droit, et celle en haut à droite (b) la conjonctive nasale de l’œil gauche. Les images du bas (c) et (d) montrent l’accumulation d’acide homogentisique sous-conjonctival (flèches noires), des cellules dendritiques (astérisques) et des zones hypo-réflectives (triangles noirs).
L’ochronose oculaire est généralement observée à partir de 30 ans et constitue souvent un signe précoce de l’AKU.
Signes oculaires fréquents
Pigmentation bleu-noir de la sclère (signe d’Osler) : survient fréquemment près du limbe cornéen nasal et temporal dans la zone de la fente palpébrale. Elle est particulièrement visible au niveau de l’insertion du muscle droit externe.
Dépôts cornéens en gouttelettes d’huile : dépôts ponctués au niveau de la membrane de Bowman. Progressent avec le temps.
Pigmentation conjonctivale : peut être associée à une dilatation des vaisseaux conjonctivaux.
Motifs de pigmentation sclérale : quatre types ont été rapportés : vermiforme, ressemblant à une pinguecula, ponctué et lamellaire.
Signes oculaires relativement rares
Astigmatisme cornéen : peut entraîner un astigmatisme progressif en raison de l’amincissement cornéen périphérique dans l’axe de la lésion.
QLa pigmentation oculaire affecte-t-elle la vision ?
A
La pigmentation de la sclère et de la conjonctive n’affecte généralement pas directement la vision. Cependant, si des dépôts cornéens progressent ou si une accumulation de pigment dans l’angle iridocornéen provoque un glaucome, une baisse de l’acuité visuelle peut survenir. Une surveillance régulière par examen ophtalmologique est recommandée.
L’AKU est causée par un déficit d’activité de l’homogentisate 1,2-dioxygénase dû à une mutation du gène HGD (3q2). Cette enzyme est principalement produite dans les hépatocytes et catalyse la dégradation de l’HGA dans la voie de dégradation de la tyrosine. Le déficit enzymatique entraîne une accumulation d’HGA dans l’organisme et une excrétion urinaire massive (1 à 8 g par jour) 1).
L’HGA se dépose dans les tissus collagènes, en particulier au niveau du nez, des oreilles, des joues, de la conjonctive, des insertions musculaires, de la cornée et de la sclère.
Dosage de l’HGA urinaire : Gold standard. Mesuré par chromatographie en phase gazeuse1)
Test d’alcalinisation des urines : Vérifier le noircissement de la couleur des urines
Test génétique HGD : Déterminer l’état homozygote ou hétérozygote composite
Biopsie des lésions oculaires : Coloration H&E montrant une dégénérescence élastosique
Si la chromatographie en phase gazeuse n’est pas disponible, le test de Benedict, le test à l’hydroxyde de sodium, le test au nitrate d’argent et le test au chlorure ferrique peuvent être utilisés en alternative1).
Il n’existait pas de traitement curatif de l’AKU depuis longtemps, mais l’apparition du nitisinone a permis un traitement modificateur de la maladie.
Nitisinone : inhibe la 4-hydroxyphénylpyruvate dioxygénase et réduit la production d’HGA. L’essai SONIA 2 (essai randomisé contrôlé multicentrique international) a montré qu’une dose de 10 mg/jour réduisait les douleurs articulaires, améliorait la densité osseuse et ralentissait la progression de la pigmentation oculaire2). En 2020, l’Agence européenne des médicaments (EMA) a approuvé son utilisation chez les patients adultes atteints d’AKU2)
Acide ascorbique (vitamine C) : fondement théorique par son action antioxydante, mais efficacité non démontrée1)2)
Régime pauvre en protéines : restriction de l’apport en phénylalanine et tyrosine. Observance à long terme difficile chez l’adulte2)
La kératopathie dendritique due à la tyrosinémie a été rapportée comme effet secondaire du nitisinone1). Le maintien du taux de tyrosine en dessous de 500-600 μmol/L permettrait de prévenir cet effet secondaire1).
Dans l’étude SONIA 2, le nitisinone à 10 mg/jour a montré une réduction des douleurs articulaires et vertébrales, une amélioration de la densité minérale osseuse (T-score) et un ralentissement de la progression de la pigmentation oculaire sur 48 mois2).
Examens ophtalmologiques réguliers : pour surveiller la progression de la pigmentation et l’apparition d’un glaucome
Correction réfractive : envisagée en cas d’astigmatisme cornéen progressif
Gestion du glaucome : en cas de glaucome associé à une accumulation de pigment dans l’angle, un traitement approprié doit être instauré
QQu'est-ce que le nitisinone ?
A
Le nitisinone est un médicament qui inhibe la 4-hydroxyphénylpyruvate dioxygénase et réduit la production d’HGA. Initialement approuvé pour le traitement de la tyrosinémie héréditaire de type 1. L’étude SONIA 1 a montré qu’une dose de 8 mg/jour réduisait l’HGA urinaire sur 24 heures de 98,8 % 2). L’étude SONIA 2 a confirmé le ralentissement de la progression des symptômes articulaires et oculaires, et le médicament a été approuvé en Europe en 2020 pour les patients adultes atteints d’AKU 2).
L’homogentisate 1,2-dioxygénase est produite dans les hépatocytes et convertit l’HGA en maléylacétoacétate dans la voie de dégradation de la tyrosine. Un déficit de cette enzyme entraîne une accumulation d’HGA dans l’organisme.
L’HGA forme des polymères de type mélanine (pigment noirâtre des tissus) par un processus d’oxydation via l’acide benzoquinone acétique. Ce pigment adhère au tissu conjonctif, provoquant une décoloration bleu-noir et des lésions tissulaires.
L’HGA exerce une toxicité directe sur les chondrocytes, entraînant une nécrose du cartilage et une destruction articulaire accélérée2). Un processus similaire se produit au niveau des valves cardiaques, et la prévalence de la sténose aortique atteint 22,2 % chez les patients atteints d’AKU2). Le taux de télopeptide N-terminal du collagène (NTx) urinaire est élevé, contribuant à une résorption osseuse accrue et à l’ostéoporose2).
L’AKU ne raccourcit pas l’espérance de vie, mais elle affecte significativement la qualité de vie en raison de l’arthropathie, des complications cardiovasculaires et des calculs rénaux 2). L’introduction précoce du nitisinone peut prévenir l’accumulation d’HGA et potentiellement inhiber la progression de l’ochronose 1)2). Cependant, l’effet du nitisinone sur l’arthropathie ochronotique déjà avancée est limité, d’où l’importance d’un diagnostic et d’un traitement précoces 1).
La pigmentation oculaire n’est pas non progressive ; elle s’étend avec l’âge. Le pronostic visuel est relativement bon en l’absence de glaucome associé.
Bhatti IA, Saqib M, Rehman IU, Amjed S, Hashim HT, Butt AA. Managing Alkaptonuria in Absence of Appropriate Medication: A Case Report and Review of Literature. Clinical medicine & research. 2024;22(2):107-111. doi:10.3121/cmr.2024.1867. PMID:39231619; PMCID:PMC11374495.
Roopnarinesingh RC, Donlon NE, Reynolds JV. Alkaptonuria: clinical manifestations and an updated approach to treatment of a rare disease. BMJ Case Rep. 2021;14:e244240.
Gupta PC, Balamurugan R, Ram J. Ocular and systemic manifestations of alkaptonuria. QJM. 2019;112(5):369. PMID: 30476261.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.