Alkaptonuria (AKU) is a rare autosomal recessive amino acid metabolism disorder caused by deficiency of homogentisate 1,2-dioxygenase, which catalyzes the breakdown of homogentisic acid (HGA), an intermediate in the tyrosine degradation pathway. This leads to accumulation of HGA in the body. The causative gene is located on chromosome 3 (3q2).
In 1902, Sir Archibald Garrod reported it as a hereditary disease and first proposed the concept of “inborn errors of metabolism.” The worldwide incidence is estimated at 1 in 250,000 to 1,000,000 people 2). It is more frequent in Slovakia and the Dominican Republic, with an incidence of about 1 in 19,000. In Japan, it is extremely rare.
AKU has a classic triad of dark urine, ochronosis, and ochronotic arthropathy. HGA forms melanin-like polymers through an oxidation process via benzoquinone acetic acid, depositing in connective tissues and causing multisystem disease. Ocular ochronosis typically appears around age 30 and is often an early sign of the disease.
QWhat is ochronosis?
A
Ochronosis is a condition in which HGA-derived melanin-like polymers deposit in connective tissues, causing a blue-black discoloration. The name derives from its color. It deposits in connective tissues throughout the body, including the sclera, ear cartilage, joint cartilage, heart valves, and kidneys. In the eye, the sclera, conjunctiva, and cornea are affected. Unlike exogenous ochronosis (skin discoloration from topical agents such as hydroquinone), AKU-related ochronosis is endogenous (hereditary).
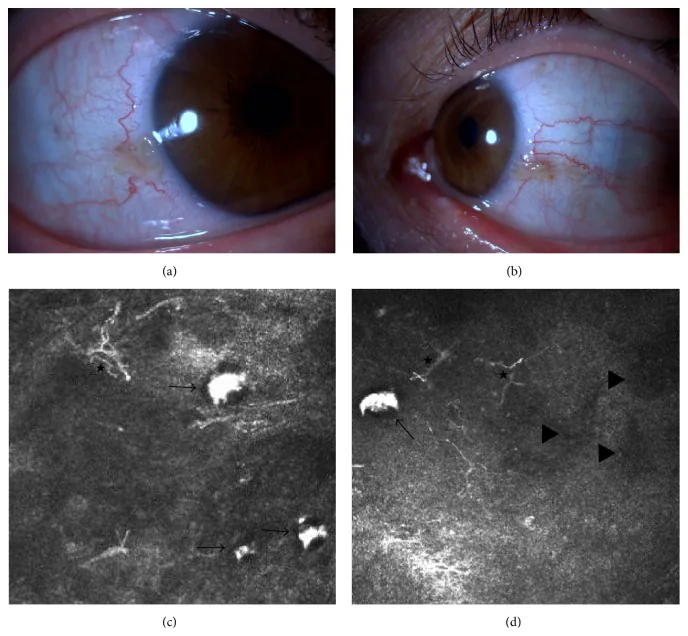
Elif Demirkilinc Biler, Suzan Guven Yilmaz, Melis Palamar, Pedram Hamrah, et al. In Vivo Confocal Microscopy and Anterior Segment Optic Coherence Tomography Findings in Ocular Ochronosis 2015 Dec 15 Case Rep Ophthalmol Med. 2015 Dec 15; 2015:592847 Figure 3. PMCID: PMC4693010. License: CC BY.
Clinical photographs and in vivo confocal microscopy images of the conjunctiva of the right and left eyes in a case of alkaptonuria (Case 2). The upper left image (a) shows the nasal conjunctiva of the right eye, and the upper right image (b) shows the nasal conjunctiva of the left eye. The lower images (c) and (d) show subconjunctival accumulation of homogentisic acid (black arrows), dendritic cells (asterisks), and hyporeflective areas (black triangles).
Ocular ochronosis is usually observed from around age 30 and is often an early sign of AKU.
Common Ocular Findings
Bluish-black scleral pigmentation (Osler’s sign): Commonly occurs near the corneal limbus on the nasal and temporal sides of the palpebral fissure. It is particularly noticeable at the insertion of the lateral rectus muscle.
Oil-drop corneal deposits: Spotty deposits at the level of Bowman’s membrane. Progresses over time.
Conjunctival pigmentation: May be accompanied by dilated conjunctival vessels.
Scleral pigmentation patterns: Four types have been reported: worm-like, pinguecula-like, punctate, and lamellar.
Relatively Rare Ocular Findings
Corneal astigmatism: Progressive astigmatism may occur due to thinning of the peripheral cornea along the axis of the lesion.
Scleral and conjunctival pigmentation itself usually does not directly affect vision. However, if corneal deposits progress or glaucoma develops due to pigment accumulation in the angle, vision loss may occur. Regular ophthalmologic follow-up is recommended.
AKU is caused by a deficiency in homogentisate 1,2-dioxygenase activity due to mutations in the HGD gene (3q2). This enzyme is mainly produced in liver cells and catalyzes the breakdown of HGA in the tyrosine degradation pathway. Enzyme deficiency leads to accumulation of HGA in the body and massive excretion in urine (1–8 g per day)1).
HGA deposits in collagen tissues, particularly in the nose, ears, cheeks, conjunctiva, muscle attachments, cornea, and sclera.
Urinary HGA quantification: Gold standard. Measured by gas chromatography1).
Urine alkalinization test: Confirms darkening of urine color
HGD gene testing: Determines homozygous or compound heterozygous status
Biopsy of ocular lesions: H&E staining shows elastosis-like degeneration
If gas chromatography is not available, Benedict’s test, sodium hydroxide test, silver nitrate test, or ferric chloride test can be used as alternatives1).
For a long time, there was no curative treatment for AKU, but the advent of nitisinone has made disease-modifying therapy possible.
Nitisinone: Inhibits 4-hydroxyphenylpyruvate dioxygenase, reducing HGA production. The SONIA 2 trial (international multicenter randomized controlled trial) showed that a dose of 10 mg/day reduced joint pain, improved bone density, and slowed the progression of ocular pigmentation2). In 2020, the European Medicines Agency (EMA) approved its use in adult AKU patients2)
Ascorbic acid (vitamin C): Has a theoretical basis due to antioxidant effects, but efficacy has not been demonstrated1)2)
Low-protein diet: Restricts intake of phenylalanine and tyrosine. Difficult for adults to adhere to long-term2)
Side effects of nitisinone include dendritic keratopathy due to tyrosinemia1). Maintaining tyrosine levels below 500–600 μmol/L is thought to prevent side effects1).
In the SONIA 2 trial, nitisinone 10 mg/day demonstrated reductions in joint and spinal pain, improvement in bone density (T-score), and suppression of progression of ocular pigmentation over 48 months 2).
Regular eye examinations: To monitor progression of pigmentation and development of glaucoma
Refractive correction: Considered when corneal astigmatism progresses
Glaucoma management: Appropriate treatment if glaucoma occurs due to angle pigment accumulation
QWhat kind of drug is nitisinone?
A
Nitisinone is a drug that inhibits 4-hydroxyphenylpyruvate dioxygenase, suppressing the production of HGA. It was originally approved for the treatment of hereditary tyrosinemia type 1. The SONIA 1 trial showed that 8 mg/day reduced 24-hour urinary HGA by 98.8% 2). The SONIA 2 trial confirmed suppression of progression of joint and ocular symptoms, and it was approved for use in adult AKU patients in Europe in 2020 2).
Homogentisate 1,2-dioxygenase is produced in hepatocytes and converts HGA to maleylacetoacetate in the tyrosine degradation pathway. Deficiency of this enzyme leads to accumulation of HGA in the body.
HGA forms melanin-like polymers (ochronotic pigment) through an oxidation process mediated by benzoquinone acetic acid. This pigment adheres to connective tissue, causing bluish-black discoloration and tissue damage.
HGA is directly toxic to chondrocytes, leading to cartilage necrosis and accelerated joint destruction2). A similar process occurs in heart valves, and the prevalence of aortic stenosis reaches 22.2% in AKU patients2). Urinary collagen N-telopeptide (NTx) is elevated, contributing to increased bone resorption and osteoporosis2).
AKU does not shorten lifespan itself, but it significantly affects quality of life due to arthropathy, cardiovascular complications, and kidney stones2). Early introduction of nitisinone can prevent HGA accumulation and potentially suppress the progression of ochronosis1)2). However, the effect of nitisinone on already advanced ochronotic arthropathy is limited, making early diagnosis and treatment important1).
Ocular pigmentation is not non-progressive; it expands over time. Visual prognosis is relatively good unless glaucoma develops.
Bhatti IA, Saqib M, Rehman IU, Amjed S, Hashim HT, Butt AA. Managing Alkaptonuria in Absence of Appropriate Medication: A Case Report and Review of Literature. Clinical medicine & research. 2024;22(2):107-111. doi:10.3121/cmr.2024.1867. PMID:39231619; PMCID:PMC11374495.
Roopnarinesingh RC, Donlon NE, Reynolds JV. Alkaptonuria: clinical manifestations and an updated approach to treatment of a rare disease. BMJ Case Rep. 2021;14:e244240.
Gupta PC, Balamurugan R, Ram J. Ocular and systemic manifestations of alkaptonuria. QJM. 2019;112(5):369. PMID: 30476261.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.