La alcaptonuria (AKU) es un trastorno raro del metabolismo de aminoácidos de herencia autosómica recesiva causado por la deficiencia de homogentisato 1,2-dioxigenasa, que cataliza la degradación del ácido homogentísico (HGA), un intermediario en la vía de degradación de la tirosina. Esto lleva a la acumulación de HGA en el cuerpo. El gen causante se localiza en el cromosoma 3 (3q2).
En 1902, Sir Archibald Garrod la reportó como una enfermedad hereditaria y propuso por primera vez el concepto de “errores innatos del metabolismo”. La incidencia mundial se estima en 1 de cada 250,000 a 1,000,000 de personas 2). Es más frecuente en Eslovaquia y la República Dominicana, con una incidencia de aproximadamente 1 en 19,000. En Japón, es extremadamente rara.
La AKU presenta una tríada clásica de orina oscura, ocronosis y artropatía ocronótica. El HGA forma polímeros similares a la melanina a través de un proceso de oxidación vía ácido benzoquinona acético, depositándose en los tejidos conectivos y causando enfermedad multisistémica. La ocronosis ocular típicamente aparece alrededor de los 30 años y a menudo es un signo temprano de la enfermedad.
Q¿Qué es la ocronosis?
A
La ocronosis es una condición en la que los polímeros similares a la melanina derivados del HGA se depositan en los tejidos conectivos, causando una decoloración azul-negra. El nombre deriva de su color. Se deposita en tejidos conectivos de todo el cuerpo, incluyendo esclerótica, cartílago auricular, cartílago articular, válvulas cardíacas y riñones. En el ojo, se afectan la esclerótica, la conjuntiva y la córnea. A diferencia de la ocronosis exógena (decoloración de la piel por agentes tópicos como la hidroquinona), la ocronosis relacionada con AKU es endógena (hereditaria).
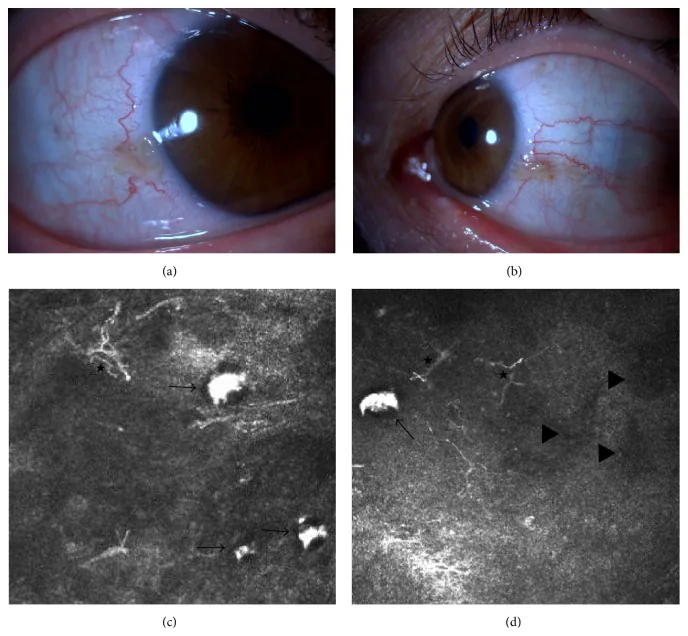
Elif Demirkilinc Biler, Suzan Guven Yilmaz, Melis Palamar, Pedram Hamrah, et al. In Vivo Confocal Microscopy and Anterior Segment Optic Coherence Tomography Findings in Ocular Ochronosis 2015 Dec 15 Case Rep Ophthalmol Med. 2015 Dec 15; 2015:592847 Figure 3. PMCID: PMC4693010. License: CC BY.
Fotografías clínicas e imágenes de microscopía confocal in vivo de la conjuntiva de los ojos derecho e izquierdo en un caso de alcaptonuria (Caso 2). La imagen superior izquierda (a) muestra la conjuntiva nasal del ojo derecho, y la imagen superior derecha (b) muestra la conjuntiva nasal del ojo izquierdo. Las imágenes inferiores (c) y (d) muestran acumulación subconjuntival de ácido homogentísico (flechas negras), células dendríticas (asteriscos) y áreas hiporreflectivas (triángulos negros).
La ocronosis ocular generalmente se observa alrededor de los 30 años y suele ser un signo temprano de AKU.
Hallazgos oculares frecuentes
Pigmentación negroazulada de la esclerótica (signo de Osler): Común cerca del limbo corneal en los lados nasal y temporal de la hendidura palpebral. Es particularmente notable en la inserción del músculo recto lateral.
Depósitos corneales en gota de aceite: Depósitos punteados a nivel de la membrana de Bowman. Progresan con el tiempo.
Pigmentación conjuntival: Puede acompañarse de vasos conjuntivales dilatados.
Patrones de pigmentación escleral: Se han descrito cuatro tipos: vermiforme, tipo pingüécula, punteado y laminar.
Hallazgos oculares relativamente raros
Astigmatismo corneal: Puede ocurrir astigmatismo progresivo debido al adelgazamiento de la córnea periférica a lo largo del eje de la lesión.
La pigmentación de la esclerótica y la conjuntiva generalmente no afecta directamente la visión. Sin embargo, si los depósitos corneales progresan o se desarrolla glaucoma debido a la acumulación de pigmento en el ángulo, puede ocurrir pérdida de visión. Se recomienda seguimiento oftalmológico regular.
La AKU es causada por una deficiencia en la actividad de la homogentisato 1,2-dioxigenasa debido a mutaciones en el gen HGD (3q2). Esta enzima se produce principalmente en las células hepáticas y cataliza la degradación del HGA en la vía de degradación de la tirosina. La deficiencia enzimática provoca la acumulación de HGA en el cuerpo y su excreción masiva en la orina (1–8 g por día)1).
El HGA se deposita en los tejidos de colágeno, especialmente en la nariz, orejas, mejillas, conjuntiva, inserciones musculares, córnea y esclerótica.
Cuantificación de HGA en orina: Estándar de oro. Se mide mediante cromatografía de gases1).
Prueba de alcalinización de la orina: Confirma el oscurecimiento del color de la orina
Prueba genética de HGD: Determina el estado homocigoto o heterocigoto compuesto
Biopsia de lesiones oculares: La tinción H&E muestra degeneración similar a elastosis
Si no se dispone de cromatografía de gases, se pueden utilizar como alternativas la prueba de Benedict, la prueba de hidróxido de sodio, la prueba de nitrato de plata o la prueba de cloruro férrico1).
Durante mucho tiempo no existió un tratamiento curativo para la AKU, pero la llegada de la nitisinona ha hecho posible la terapia modificadora de la enfermedad.
Nitisinona: Inhibe la 4-hidroxifenilpiruvato dioxigenasa, reduciendo la producción de HGA. El ensayo SONIA 2 (ensayo controlado aleatorizado multicéntrico internacional) mostró que una dosis de 10 mg/día redujo el dolor articular, mejoró la densidad ósea y retrasó la progresión de la pigmentación ocular2). En 2020, la Agencia Europea de Medicamentos (EMA) aprobó su uso en pacientes adultos con AKU2)
Ácido ascórbico (vitamina C): Tiene una base teórica debido a los efectos antioxidantes, pero no se ha demostrado su eficacia1)2)
Dieta baja en proteínas: Restringe la ingesta de fenilalanina y tirosina. Difícil de mantener a largo plazo en adultos2)
Los efectos secundarios de la nitisinona incluyen queratopatía dendrítica debida a tirosinemia1). Mantener los niveles de tirosina por debajo de 500–600 μmol/L se considera que previene los efectos secundarios1).
En el ensayo SONIA 2, nitisinona 10 mg/día demostró reducciones del dolor articular y espinal, mejora de la densidad ósea (T-score) y supresión de la progresión de la pigmentación ocular durante 48 meses 2).
Exámenes oculares regulares: Para monitorear la progresión de la pigmentación y el desarrollo de glaucoma
Corrección refractiva: Considerada cuando el astigmatismo corneal progresa
Manejo del glaucoma: Tratamiento adecuado si ocurre glaucoma debido a la acumulación de pigmento en el ángulo
Q¿Qué tipo de fármaco es la nitisinona?
A
La nitisinona es un fármaco que inhibe la 4-hidroxifenilpiruvato dioxigenasa, suprimiendo la producción de HGA. Originalmente fue aprobado para el tratamiento de la tirosinemia hereditaria tipo 1. El ensayo SONIA 1 mostró que 8 mg/día redujo el HGA urinario de 24 horas en un 98.8% 2). El ensayo SONIA 2 confirmó la supresión de la progresión de los síntomas articulares y oculares, y fue aprobado para su uso en pacientes adultos con AOU en Europa en 2020 2).
La homogentisato 1,2-dioxigenasa se produce en los hepatocitos y convierte el HGA en maleilacetoacetato en la vía de degradación de la tirosina. La deficiencia de esta enzima conduce a la acumulación de HGA en el cuerpo.
El HGA forma polímeros similares a la melanina (pigmento ocronótico) a través de un proceso de oxidación mediado por el ácido benzoquinona acético. Este pigmento se adhiere al tejido conectivo, causando decoloración azul-negruzca y daño tisular.
El HGA es directamente tóxico para los condrocitos, lo que lleva a necrosis del cartílago y destrucción articular acelerada2). Un proceso similar ocurre en las válvulas cardíacas, y la prevalencia de estenosis aórtica alcanza el 22.2% en pacientes con AKU2). El telopéptido N-terminal del colágeno (NTx) en orina está elevado, contribuyendo al aumento de la resorción ósea y la osteoporosis2).
La AKU no acorta la esperanza de vida por sí misma, pero afecta significativamente la calidad de vida debido a artropatía, complicaciones cardiovasculares y cálculos renales2). La introducción temprana de nitisinona puede prevenir la acumulación de HGA y potencialmente suprimir la progresión de la ocronosis1)2). Sin embargo, el efecto de la nitisinona en la artropatía ocronótica ya avanzada es limitado, por lo que el diagnóstico y tratamiento tempranos son importantes1).
La pigmentación ocular no es no progresiva; se expande con el tiempo. El pronóstico visual es relativamente bueno a menos que se desarrolle glaucoma.
Bhatti IA, Saqib M, Rehman IU, Amjed S, Hashim HT, Butt AA. Managing Alkaptonuria in Absence of Appropriate Medication: A Case Report and Review of Literature. Clinical medicine & research. 2024;22(2):107-111. doi:10.3121/cmr.2024.1867. PMID:39231619; PMCID:PMC11374495.
Roopnarinesingh RC, Donlon NE, Reynolds JV. Alkaptonuria: clinical manifestations and an updated approach to treatment of a rare disease. BMJ Case Rep. 2021;14:e244240.
Gupta PC, Balamurugan R, Ram J. Ocular and systemic manifestations of alkaptonuria. QJM. 2019;112(5):369. PMID: 30476261.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.