L’alcaptonuria (AKU) è una rara malattia autosomica recessiva del metabolismo degli amminoacidi causata da un deficit dell’enzima omogentisato 1,2-diossigenasi, che catalizza la degradazione dell’acido omogentisico (HGA), un intermedio della via di degradazione della tirosina, portando all’accumulo di HGA nell’organismo. Il gene responsabile si trova sul cromosoma 3 (3q2).
Nel 1902, Sir Archibald Garrod la descrisse come malattia ereditaria, proponendo per primo il concetto di “errori congeniti del metabolismo”. L’incidenza mondiale è stimata in 1 su 250.000-1.000.000 di persone 2). In Slovacchia e Repubblica Dominicana è più frequente, circa 1 su 19.000 persone. In Giappone è una malattia estremamente rara.
L’AKU è classicamente caratterizzata dalla triade di urine scure, ocronosi e artropatia ocronotica. L’HGA forma polimeri simili alla melanina attraverso un processo di ossidazione mediato dall’acido benzochinonacetico, che si depositano nei tessuti connettivi causando una malattia multisistemica. L’ocronosi oculare compare in media intorno ai 30 anni ed è spesso un segno precoce della malattia.
QCos'è l'ocronosi?
A
L’ocronosi è una condizione in cui i polimeri simili alla melanina derivati dall’HGA si depositano nei tessuti connettivi, causando una colorazione blu-nera. Il nome deriva da questa colorazione. I depositi si verificano in tutto il corpo, inclusi sclera, cartilagine auricolare, cartilagine articolare, valvole cardiache e reni. Nell’occhio sono colpiti sclera, congiuntiva e cornea. A differenza dell’ocronosi esogena (causata da farmaci topici come l’idrochinone), l’ocronosi dell’AKU è endogena (ereditaria).
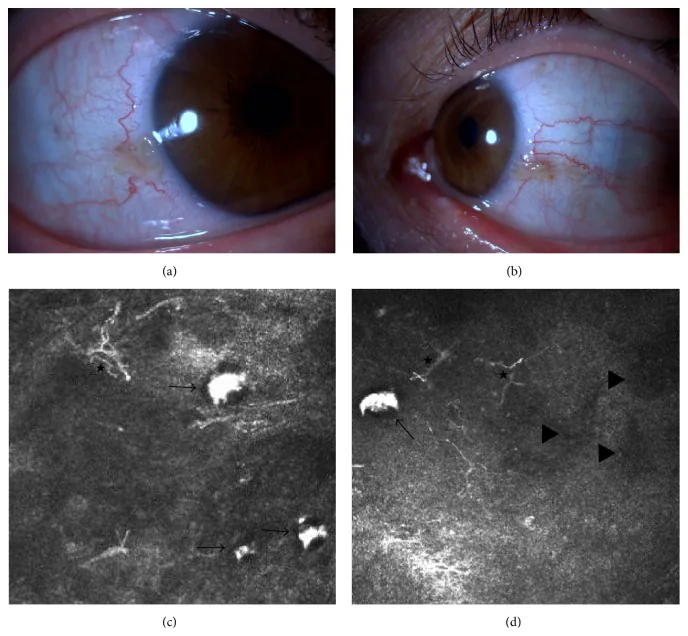
Elif Demirkilinc Biler, Suzan Guven Yilmaz, Melis Palamar, Pedram Hamrah, et al. In Vivo Confocal Microscopy and Anterior Segment Optic Coherence Tomography Findings in Ocular Ochronosis 2015 Dec 15 Case Rep Ophthalmol Med. 2015 Dec 15; 2015:592847 Figure 3. PMCID: PMC4693010. License: CC BY.
Fotografie cliniche e immagini di microscopia confocale in vivo della congiuntiva dell’occhio destro e sinistro in un caso di alcaptonuria. L’immagine in alto a sinistra (a) mostra la congiuntiva nasale dell’occhio destro, quella in alto a destra (b) la congiuntiva nasale dell’occhio sinistro. Le immagini inferiori (c) e (d) mostrano accumulo di acido omogentisico sotto la congiuntiva (freccia nera), cellule dendritiche (asterisco) e aree iporiflettenti (triangolo nero).
L’ocronosi oculare si osserva solitamente intorno ai 30 anni e spesso rappresenta il primo segno di AKU.
Reperti oculari frequenti
Pigmentazione blu-nera della sclera (segno di Osler): si manifesta frequentemente vicino al limbo corneale nasale e temporale nella fessura palpebrale. È particolarmente visibile in corrispondenza dell’inserzione del muscolo retto laterale.
Depositi corneali a goccia d’olio: depositi punteggiati a livello della membrana di Bowman. Progressivi nel tempo.
Pigmentazione congiuntivale: può essere associata a vasi congiuntivali dilatati.
Pattern di pigmentazione sclerale: sono stati descritti quattro tipi: vermiforme, pinguecola-simile, puntiforme e laminare.
Reperti oculari relativamente rari
Astigmatismo corneale: può causare astigmatismo progressivo a causa dell’assottigliamento corneale periferico lungo l’asse della lesione.
La pigmentazione della sclera e della congiuntiva di solito non influisce direttamente sulla vista. Tuttavia, se la pigmentazione progredisce sulla cornea o se l’accumulo di pigmento nell’angolo della camera anteriore causa glaucoma, può portare a una riduzione della vista. Si raccomanda un monitoraggio regolare tramite visite oculistiche periodiche.
L’AKU è causata da una mutazione nel gene HGD (3q2) che porta a una carenza di attività dell’enzima omogentisato 1,2-diossigenasi. Questo enzima è prodotto principalmente negli epatociti e catalizza la degradazione dell’acido omogentisico (HGA) nella via di degradazione della tirosina. La carenza enzimatica porta all’accumulo di HGA nell’organismo e alla sua massiccia escrezione nelle urine (1-8 g al giorno) 1).
L’HGA si deposita nei tessuti collagenici, in particolare nel naso, nelle orecchie, nelle guance, nella congiuntiva, nei punti di inserzione muscolare, nella cornea e nella sclera.
Dosaggio dell’HGA urinario: gold standard. Misurato mediante gascromatografia 1)
Test di alcalinizzazione delle urine: verifica dell’oscuramento del colore delle urine
Test genetico HGD: determina lo stato omozigote o eterozigote composto
Biopsia delle lesioni oculari: alla colorazione H&E si osserva degenerazione simil-elastosi
Se la gascromatografia non è disponibile, possono essere utilizzati il test di Benedict, il test con idrossido di sodio, il test con nitrato d’argento o il test con cloruro ferrico 1).
Per l’AKU non esisteva a lungo una terapia curativa, ma con l’avvento della nitisinone è diventata possibile una terapia modificante la malattia.
Nitisinone: inibisce la 4-idrossifenilpiruvato diossigenasi, riducendo la produzione di HGA. Lo studio SONIA 2 (studio randomizzato controllato multicentrico internazionale) ha mostrato che una dose di 10 mg/die riduce il dolore articolare, migliora la densità ossea e rallenta la progressione della pigmentazione oculare2). Nel 2020 l’Agenzia Europea per i Medicinali (EMA) ne ha approvato l’uso nei pazienti adulti con AKU2)
Acido ascorbico (vitamina C): ha una base teorica per l’azione antiossidante, ma l’efficacia non è stata dimostrata1)2)
Dieta ipoproteica: restrizione dell’assunzione di fenilalanina e tirosina. Difficile da mantenere a lungo termine negli adulti2)
Come effetto collaterale della nitisinone è stata riportata la cheratopatia dendritica dovuta a tirosinemia 1). Si ritiene che mantenere i livelli di tirosina al di sotto di 500-600 μmol/L possa prevenire gli effetti collaterali 1).
Nello studio SONIA 2, la nitisinone 10 mg/die ha mostrato una riduzione del dolore articolare e spinale, un miglioramento della densità ossea (T-score) e un rallentamento della progressione della pigmentazione oculare per 48 mesi 2).
Visite oculistiche regolari: per monitorare la progressione della pigmentazione e l’insorgenza di glaucoma
Correzione refrattiva: da considerare in caso di astigmatismo corneale progressivo
Gestione del glaucoma: se si verifica glaucoma associato all’accumulo di pigmento nell’angolo, eseguire un trattamento appropriato
QChe tipo di farmaco è la nitisinone?
A
La nitisinone è un farmaco che inibisce la 4-idrossifenilpiruvato diossigenasi, sopprimendo la produzione di HGA. Originariamente approvato per il trattamento della tirosinemia ereditaria di tipo 1. Lo studio SONIA 1 ha mostrato che una dose di 8 mg/die riduce l’HGA urinario delle 24 ore del 98,8% 2). Lo studio SONIA 2 ha confermato la progressione rallentata dei sintomi articolari e oculari, portando all’approvazione in Europa nel 2020 per l’uso in pazienti adulti con AKU 2).
6. Fisiopatologia e meccanismo dettagliato di insorgenza
L’omogentisato 1,2-diossigenasi è prodotta negli epatociti e converte l’HGA in maleilacetoacetato nella via di degradazione della tirosina. La carenza di questo enzima porta all’accumulo di HGA nell’organismo.
L’HGA forma polimeri simili alla melanina (pigmento di annerimento tissutale) attraverso un processo di ossidazione mediato dall’acido benzochinonacetico. Questo pigmento aderisce al tessuto connettivo, causando una colorazione blu-nera e danni tissutali.
L’HGA mostra tossicità diretta verso i condrociti, portando a necrosi della cartilagine e distruzione articolare accelerata2). Un processo simile si verifica anche nelle valvole cardiache, e la prevalenza della stenosi aortica raggiunge il 22,2% nei pazienti con AKU2). I livelli urinari di N-telopeptide del collagene (NTx) sono elevati, contribuendo all’aumento del riassorbimento osseo e all’osteoporosi2).
L’AKU non riduce di per sé l’aspettativa di vita, ma ha un impatto significativo sulla qualità della vita a causa di artropatia, complicanze cardiovascolari e calcoli renali 2). L’introduzione precoce di nitisinone può prevenire l’accumulo di HGA e potenzialmente rallentare la progressione dell’ocronosi 1)2). Tuttavia, l’effetto del nitisinone sull’artropatia ocronotica già avanzata è limitato, pertanto la diagnosi e il trattamento precoci sono importanti 1).
La pigmentazione oculare non è non progressiva, ma si estende nel tempo. La prognosi visiva è relativamente buona, a meno che non sia complicata da glaucoma.
Bhatti IA, Saqib M, Rehman IU, Amjed S, Hashim HT, Butt AA. Managing Alkaptonuria in Absence of Appropriate Medication: A Case Report and Review of Literature. Clinical medicine & research. 2024;22(2):107-111. doi:10.3121/cmr.2024.1867. PMID:39231619; PMCID:PMC11374495.
Roopnarinesingh RC, Donlon NE, Reynolds JV. Alkaptonuria: clinical manifestations and an updated approach to treatment of a rare disease. BMJ Case Rep. 2021;14:e244240.
Gupta PC, Balamurugan R, Ram J. Ocular and systemic manifestations of alkaptonuria. QJM. 2019;112(5):369. PMID: 30476261.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.