A neoplasia endócrina múltipla (MEN) é um grupo raro de doenças hereditárias que aumentam o risco de desenvolvimento de neoplasias em duas ou mais glândulas endócrinas. Segue um padrão de herança autossômica dominante, com alta penetrância e expressividade variável.
A MEN é classificada principalmente em quatro tipos.
Tipo MEN1
Gene responsável: gene MEN1 (11q13)
Combinação de tumores: tumor de paratireoide, tumor hipofisário, tumor gastroenteropancreático
Prevalência: 2 a 20 pessoas por 100.000 habitantes
Características oftalmológicas: Compressão do quiasma óptico por tumor hipofisário (hemianopsia bitemporal)
MEN tipo 2 (2A e 2B)
Gene responsável: Proto-oncogene RET (10q11.2)
Combinação de tumores: carcinoma medular da tireoide (CMT), feocromocitoma, hiperparatireoidismo (apenas tipo 2A)
Prevalência: 1 em 35.000 pessoas (para MEN2 como um todo)
Características oftalmológicas: espessamento proeminente dos nervos corneanos, neuromas conjuntivais e palpebrais (especialmente no tipo MEN2B)
As características oftalmológicas da MEN são classicamente descritas no tipo MEN2B. No tipo MEN2B, além do carcinoma medular da tireoide e feocromocitoma, ocorrem neuromas mucosos e ganglioneuromatose intestinal. Os neuromas mucosos estão presentes em quase 100% dos pacientes com MEN2B e são considerados um achado patognomônico desta síndrome. Os achados oftalmológicos podem ser um indício para o diagnóstico precoce do tipo MEN2B5).
No estudo de grande coorte DutchMEN1 Study (323 casos), foi demonstrado que 38,1% dos pacientes com MEN1 tipo 1 desenvolvem tumores hipofisários 3). O prolactinoma é o tumor hipofisário mais comum e responde bem à terapia medicamentosa com agonistas dopaminérgicos.
QQuando os achados oftalmológicos do tipo MEN2B são geralmente reconhecidos?
A
Os achados característicos do tipo MEN2B podem ser reconhecidos desde a infância. O espessamento dos nervos corneanos e neuromas mucosos aparecem precocemente e podem ser os primeiros indícios clínicos do tipo MEN2B. O CMT do tipo MEN2B pode se manifestar na infância, e o reconhecimento precoce dos achados oftalmológicos determina o momento do diagnóstico e do tratamento preventivo.
Nervos corneanos proeminentes são o achado oftalmológico mais característico na MEN2B. O exame com lâmpada de fenda revela fibras nervosas espessadas dentro da córnea. Histopatologicamente, são nervos amielínicos com células de Schwann, e os axônios apresentam aparência normal. Na MEN2A, de acordo com o relato de Kinoshita et al., 16 de 28 olhos (57%) apresentaram espessamento do nervo corneano de grau 2 ou superior, e cerca de 29% apresentaram espessamento acentuado de grau 3 a 4, sendo um achado oftalmológico importante em toda a MEN1).
Neuromas conjuntivais e palpebrais são reconhecidos como proliferações nervosas espessadas, sem cápsula. Neuromas mucosos semelhantes também aparecem nos lábios, língua e mucosa jugal. Na microscopia confocal in vivo (IVCM), observam-se neuromas conjuntivais como feixes nervosos grandes e espessos com arranjo irregular, alças, ramificações e dilatações de até 1 mm; na córnea, observa-se espessamento do plexo nervoso subbasal6). Em crianças, foi relatado que neuromas mucosos perilimbais causam glaucoma secundário de ângulo aberto, necessitando de controle da pressão intraocular2).
No MEN tipo 1, a síndrome quiasmática devido a tumor hipofisário é o principal problema oftalmológico. A hemianopsia bitemporal com limite na linha média vertical é o achado mais frequente no exame de campo visual. Pode haver atrofia do nervo óptico.
Características não oftalmológicas (MEN tipo 2B) incluem lábios hipertrofiados, hábito corporal semelhante à síndrome de Marfan e ganglioneuromatose intestinal (que pode causar megacólon).
QSe o nervo corneano parecer espesso, devemos sempre suspeitar de MEN?
A
A proeminência dos nervos corneanos não é um achado específico do MEN2B. Também é observada na síndrome de Refsum, hanseníase, síndrome de Riley-Day, neurofibromatose e ictiose congênita. Além disso, doenças da córnea como ceratocone, ceratite herpética simples e distrofia polimorfa posterior da córnea podem apresentar nervos corneanos proeminentes. No entanto, geralmente não são tão acentuados como no MEN2B. O diagnóstico é feito pela combinação com outros achados sistêmicos (hipertrofia labial, hábito marfanoide, etc.).
A síndrome MEN é herdada de forma autossômica dominante. Indivíduos com mutação no RET têm 50% de risco de transmiti-la aos descendentes. Também existem casos esporádicos devido a mutações somáticas.
A causa é a mutação no gene MEN1 (cromossomo 11q13). O gene MEN1 é um gene supressor de tumor que codifica a proteína menina. A perda de função ocorre por “dois hits” com mutações em ambos os alelos, levando à proliferação celular descontrolada.
A causa são mutações de sentido trocado no proto-oncogene RET (cromossomo 10q11.2). A proteína RET é um receptor tirosina quinase envolvido na diferenciação e migração de tecidos neuroendócrinos. As mutações resultam em ganho de função anormal, levando ao desenvolvimento de tumores.
No MEN2B, cerca de 95% dos pacientes apresentam mutação germinativa no códon 918 (M918T)4). Pacientes com essa mutação desenvolvem CMT desde a infância, sendo a forma mais agressiva. O diagnóstico de MEN2B costuma ser tardio; no relato de Nagaoka et al. no Japão, todos os 4 casos apresentavam neuromas mucosos e sintomas gastrointestinais desde a infância, mas foram diagnosticados apenas no estágio de CMT sintomático5).
Mehtab Ahmad, Imran Rizvi, Amit Jain, Noorin Zaidi Painful Hip Leading to the Diagnosis of MEN 2B Syndrome 2012 Nov 26 Case Rep Endocrinol. 2012 Nov 26; 2012:567060 Figure 2. PMCID: PMC3513729. License: CC BY.
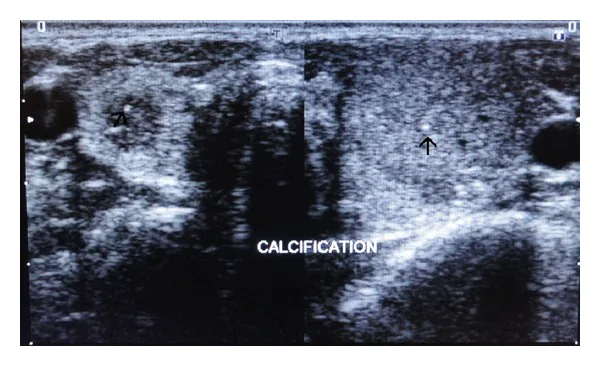
Imagens ultrassonográficas da tireoide direita e esquerda mostrando calcificações puntiformes hiperecogênicas no parênquima, indicadas por setas. Esta é uma imagem de avaliação de lesões tireoidianas relevantes na neoplasia endócrina múltipla.
O diagnóstico da síndrome MEN geralmente é feito com base na história familiar e em achados não oftalmológicos. O exame oftalmológico é particularmente útil na investigação de novos casos.
Exame com lâmpada de fenda: essencial para observar espessamento dos nervos corneanos e neuromas conjuntivais e palpebrais.
Microscopia confocal in vivo (IVCM):Permite visualizar de forma não invasiva os feixes nervosos espessados característicos do neuroma conjuntival, estruturas em alça e espessamento do plexo nervoso subbasal da córnea6)
Perimetria de Goldmann (GVF):Utilizada para avaliar a síndrome quiasmática causada por tumor hipofisário no MEN1. A hemianopsia bitemporal é o achado mais frequente
Teste de visão de cores de Ishihara:Utilizado para avaliar distúrbios de visão de cores causados por tumor hipofisário
Tomografia de coerência óptica de alta resolução (HR-OCT):Utilizada para avaliação não invasiva do neuroma conjuntival, sendo útil no diagnóstico diferencial com outras doenças
O aconselhamento genético deve ser realizado antes do teste do gene RET. Em famílias com MEN2, a triagem deve ser feita ao nascimento ou o mais precocemente possível. Todos os parentes de primeiro grau de um caso índice com diagnóstico recente de mutação no RET devem receber aconselhamento genético.
Em pacientes com mutações germinativas nos códons 883, 918 ou 922 do MEN2B, a tireoidectomia total com dissecção dos linfonodos da região central deve ser realizada nos primeiros meses de vida, devido à penetrância vitalícia do CMT de quase 100%.
O oftalmologista desempenha um papel importante na detecção precoce das manifestações oftalmológicas na NEM, no encaminhamento adequado e na reabilitação visual. Nos tumores hipofisários da NEM tipo 1, são necessários exames regulares de campo visual e avaliação da acuidade visual. Na NEM tipo 2B, deve-se realizar o manejo do olho seco e a medição regular da pressão intraocular.
QQuando diagnosticado com NEM tipo 2B, qual acompanhamento oftalmológico é necessário?
A
No tipo MEN2B, exames regulares com lâmpada de fenda observam alterações nos nervos corneanos e neuromas conjuntivais/palpebrais. Verifique a presença de sintomas de olho seco e, se necessário, maneje com lágrimas artificiais. Devido ao risco de glaucoma de ângulo aberto, recomenda-se medição regular da pressão intraocular e exames de campo visual. Além disso, avalie por imagem a possível presença de tumores hipofisários.
6. Fisiopatologia e mecanismos detalhados de desenvolvimento
O tipo MEN1 se desenvolve pela perda de função de genes supressores tumorais com base na “hipótese dos dois hits”. A primeira mutação (mutação germinativa) é herdada geneticamente, e a segunda mutação (mutação somática) é adquirida posteriormente, resultando na perda completa da função da proteína menina e na desregulação do controle da proliferação celular.
A menina é expressa tanto em tecidos endócrinos quanto não endócrinos, mas o mecanismo exato que leva à carcinogênese ainda não foi elucidado. Tumores hipofisários que comprimem o quiasma óptico causam hemianopsia bitemporal.
A proteína RET é um receptor tirosina quinase envolvido na diferenciação e migração de tecidos neuroendócrinos em desenvolvimento. Mutações de sentido trocado no gene RET resultam em substituição de um único aminoácido, causando ativação constitutiva (ganho de função) da proteína.
O RET é expresso em múltiplos tecidos, incluindo células parafoliculares da tireoide (células C), paratireoides, gânglios entéricos, células cromafins da suprarrenal e sistemas nervosos periférico e central. Esse padrão de expressão amplo explica os diversos padrões tumorais no MEN tipo 2.
O exame histopatológico dos nervos corneanos espessados no tipo MEN2B mostra nervos amielínicos com células de Schwann. Os axônios têm aparência normal e variam em diâmetro entre 0,1 e 1,4 nm. Os neuromas conjuntivais e palpebrais são observados como proliferações nervosas sem cápsula.
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. American journal of ophthalmology. 1991;111(3):307-11. doi:10.1016/s0002-9394(14)72314-1. PMID:1672053.
Chang TC, Okafor KC, Cavuoto KM, Dubovy SR, Karp CL. Pediatric Multiple Endocrine Neoplasia Type 2B: Clinicopathological Correlation of Perilimbal Mucosal Neuromas and Treatment of Secondary Open-Angle Glaucoma. Ocular oncology and pathology. 2018;4(3):196-198. doi:10.1159/000484053. PMID:29765955; PMCID:PMC5939668.
de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab. 2015;100(9):3288-3296. doi:10.1210/jc.2015-2015.
Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610. doi:10.1089/thy.2014.0335. PMID:25810047; PMCID:PMC4490627.
Nagaoka R, Sugitani I, Sanada M, Jikuzono T, Okamura R, Igarashi T, et al. The Reality of Multiple Endocrine Neoplasia Type 2B Diagnosis: Awareness of Unique Physical Appearance Is Important. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2018;85(3):178-182. doi:10.1272/jnms.JNMS.2018_85-26. PMID:30135345.
Lam D, Villaret J, Nguyen Kim P, Gabison E, Cochereau I, Doan S. In Vivo Confocal Microscopy of Prominent Conjunctival and Corneal Nerves in Multiple Endocrine Neoplasia Type 2B. Cornea. 2019;38(11):1453-1455. doi:10.1097/ICO.0000000000002028. PMID:31205161.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.