Die multiple endokrine Neoplasie (MEN) ist eine seltene Gruppe erblicher Erkrankungen, bei denen das Risiko für Neoplasien in zwei oder mehr endokrinen Drüsen erhöht ist. Sie folgt einem autosomal-dominanten Erbgang mit hoher Penetranz und variabler Expressivität.
Prävalenz : 2 bis 20 Personen pro 100.000 Einwohner
Ophthalmologische Merkmale: Kompression des Chiasma opticum durch Hypophysentumor (bitemporale Hemianopsie)
MEN Typ 2 (2A und 2B)
Verantwortliches Gen: RET-Proto-Onkogen (10q11.2)
Tumorkombination: Medulläres Schilddrüsenkarzinom (MTC), Phäochromozytom, Hyperparathyreoidismus (nur bei Typ 2A)
Prävalenz: 1 von 35.000 Personen (für MEN Typ 2 insgesamt)
Ophthalmologische Merkmale: deutliche Verdickung der Hornhautnerven, konjunktivale und lidständige Neurome (besonders bei MEN2B)
Die ophthalmologischen Merkmale des MEN werden klassischerweise beim MEN2B beschrieben. MEN2B umfasst neben dem medullären Schilddrüsenkarzinom und Phäochromozytom auch Schleimhautneurome und intestinale Ganglioneuromatose. Schleimhautneurome treten bei nahezu 100 % der MEN2B-Patienten auf und gelten als pathognomonisch für dieses Syndrom. Ophthalmologische Befunde können ein früher Hinweis auf MEN2B sein 5).
Beim MEN1 zeigte die DutchMEN1-Studie an einer großen Kohorte (323 Fälle), dass 38,1 % der Patienten einen Hypophysentumor entwickeln 3). Prolaktinome sind die häufigsten Hypophysentumoren und sprechen gut auf eine medikamentöse Therapie mit Dopaminagonisten an.
QAb welchem Alter sind die ophthalmologischen Befunde des MEN2B erkennbar?
A
Die charakteristischen Befunde des MEN2B sind bereits im Kindesalter erkennbar. Die Verdickung der Hornhautnerven und Schleimhautneurome treten früh auf und können der erste klinische Hinweis auf MEN2B sein. Das MTC bei MEN2B kann bereits im Säuglingsalter auftreten, und die frühzeitige Erkennung der ophthalmologischen Befunde beeinflusst den Zeitpunkt der Diagnose und der prophylaktischen Behandlung.
Prominente Korneanerven sind der charakteristischste ophthalmologische Befund bei MEN2B. Die Spaltlampenmikroskopie zeigt verdickte Nervenfasern in der Hornhaut. Histopathologisch handelt es sich um marklose Nerven mit Schwann-Zellen, die Axone weisen ein normales Erscheinungsbild auf. Auch bei MEN2A wurde laut Kinoshita et al. bei 16 von 28 Augen (57%) eine Korneanervenverdickung Grad 2 oder höher festgestellt, und bei etwa 29% wurde eine deutliche Verdickung Grad 3–4 beobachtet, was einen wichtigen ophthalmologischen Befund bei MEN insgesamt darstellt 1).
Konjunktivale und Lid-Neurome zeigen sich als kapsellose, verdickte Nervenproliferationen. Ähnliche Schleimhautneurome treten auch an Lippen, Zunge und Wangenschleimhaut auf. In der In-vivo-Konfokalmikroskopie (IVCM) zeigen sich bei konjunktivalen Neuromen große, dicke Nervenbündel mit unregelmäßiger Anordnung, Schlingen, Verzweigungen und Erweiterungen bis zu 1 mm, während in der Hornhaut eine Verdickung des subbasalen Nervenplexus beobachtet wird 6). Bei Kindern wurde berichtet, dass perlimbale Schleimhautneurome ein sekundäres Offenwinkelglaukom verursachen können, das eine Augeninnendruckkontrolle erfordert 2).
Bei MEN1 ist das Chiasma-Syndrom aufgrund eines Hypophysentumors das Hauptproblem. In der Gesichtsfelduntersuchung tritt am häufigsten eine bitemporale Hemianopsie mit vertikaler Mittellinienbegrenzung auf. Sie kann von einer Optikusatrophie begleitet sein.
Nicht-ophthalmologische Merkmale (MEN2B) umfassen Lippenhypertrophie, marfanoiden Habitus und intestinale Ganglioneuromatose (die zu einem Megakolon führen kann).
QSollte bei verdickten Korneanerven immer ein MEN vermutet werden?
A
Die Prominenz der Korneanerven ist nicht spezifisch für MEN2B. Sie tritt auch beim Refsum-Syndrom, bei Lepra, beim Riley-Day-Syndrom, bei Neurofibromatose und bei kongenitaler Ichthyose auf. Auch bei Hornhauterkrankungen wie Keratokonus, Herpes-simplex-Keratitis und posteriorer polymorpher Hornhautdystrophie können die Korneanerven auffällig sein. Allerdings sind sie in der Regel nicht so ausgeprägt wie bei MEN2B. Die Diagnose wird in Kombination mit anderen systemischen Befunden (Lippenhypertrophie, marfanoider Habitus usw.) gestellt.
Das MEN-Syndrom wird autosomal-dominant vererbt. Personen mit einer RET-Mutation haben ein Vererbungsrisiko von 50% für ihre Nachkommen. Es gibt auch sporadische Fälle durch somatische Mutationen.
Ursache ist eine Mutation des MEN1-Gens (Chromosom 11q13). Das MEN1-Gen kodiert für Menin, ein Tumorsuppressorprotein. Durch einen „Two-Hit“-Verlust beider Allele kommt es zum Funktionsverlust und unkontrolliertem Zellwachstum.
Ursache ist eine Missense-Mutation des RET-Protoonkogens (Chromosom 10q11.2). Das RET-Protein ist ein Tyrosinkinase-Rezeptor, der an der Differenzierung und Migration neuroendokriner Gewebe beteiligt ist. Die Mutation führt zu einem anormalen Funktionsgewinn und damit zur Tumorentstehung.
Beim MEN2B-Typ weisen etwa 95 % der Patienten eine Keimbahnmutation am Codon 918 (M918T-Mutation) auf 4). Patienten mit dieser Mutation erkranken bereits im Säuglingsalter und zeigen das aggressivste MTC. Andererseits wird die Diagnose bei MEN2B-Patienten oft verzögert; in einer japanischen Fallserie von Nagaoka et al. wurden alle 4 Fälle trotz Schleimhautneuromen und gastrointestinalen Symptomen ab dem Säuglingsalter erst im Stadium des symptomatischen MTC diagnostiziert 5).
Mehtab Ahmad, Imran Rizvi, Amit Jain, Noorin Zaidi Painful Hip Leading to the Diagnosis of MEN 2B Syndrome 2012 Nov 26 Case Rep Endocrinol. 2012 Nov 26; 2012:567060 Figure 2. PMCID: PMC3513729. License: CC BY.
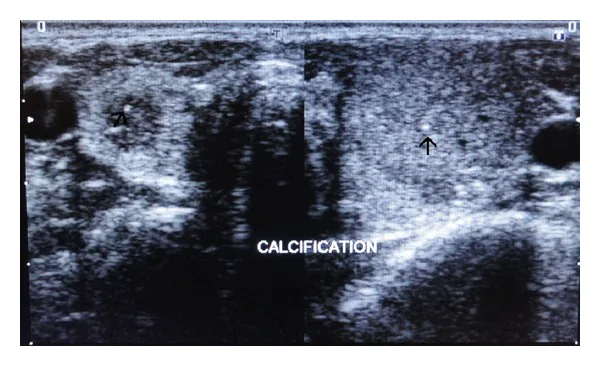
Ultraschallbilder des rechten und linken Schilddrüsenlappens, die punktförmige hyperechogene Verkalkungen im Parenchym zeigen (durch Pfeile markiert). Dies ist ein Bewertungsbild von Schilddrüsenläsionen, die bei der multiplen endokrinen Neoplasie problematisch sind.
Die Diagnose des MEN-Syndroms basiert in der Regel auf der Familienanamnese und nicht-ophthalmologischen Befunden. Die augenärztliche Untersuchung ist besonders bei der Abklärung von Neuerkrankungen nützlich.
Spaltlampenmikroskopie: unerlässlich zur Beobachtung von Hornhautnervenverdickungen, konjunktivalen und lidständigen Neuromen
In-vivo-Konfokalmikroskopie (IVCM): ermöglicht die nicht-invasive Visualisierung der charakteristischen großen Nervenbündel und Schlingenstrukturen konjunktivaler Neurome sowie der Verdickung des subbasalen Hornhautnervengeflechts 6)
Goldmann-Perimeter (GVF): wird zur Beurteilung des Chiasmasyndroms aufgrund eines Hypophysentumors bei MEN1 verwendet. Die bitemporale Hemianopsie ist der häufigste Befund.
Ishihara-Farbsehtest: wird zur Beurteilung von Farbsehstörungen aufgrund eines Hypophysentumors verwendet
Hochauflösende optische Kohärenztomographie (HR-OCT): wird zur nicht-invasiven Beurteilung konjunktivaler Neurome verwendet und ist nützlich zur Abgrenzung von anderen Erkrankungen
Die Behandlung von MEN-Patienten erfordert eine multidisziplinäre Zusammenarbeit, an der klinische Genetiker, Endokrinologen, Chirurgen und Onkologen beteiligt sind.
Die genetische Beratung sollte vor dem RET-Gentest erfolgen. Bei Familien mit MEN2 sollte das Screening bei der Geburt oder so früh wie möglich durchgeführt werden. Allen erstgradigen Verwandten von neu diagnostizierten Indexpatienten mit RET-Mutation sollte eine genetische Beratung angeboten werden.
Bei Patienten mit MEN2B und Keimbahnmutationen in den Codons 883, 918 oder 922 sollte innerhalb der ersten Lebensmonate eine totale Thyreoidektomie mit zentraler Lymphknotendissektion durchgeführt werden, da die lebenslange Penetranz des MTC nahezu 100% beträgt.
Der Augenarzt spielt eine wichtige Rolle bei der Früherkennung ophthalmologischer Manifestationen bei MEN, der angemessenen Überweisung und der Sehrehabilitation. Bei Hypophysentumoren im Rahmen von MEN1 sind regelmäßige Gesichtsfelduntersuchungen und Sehschärfebewertungen erforderlich. Bei MEN2B erfolgt die Behandlung von trockenen Augen und regelmäßige Augeninnendruckmessungen.
QWelche ophthalmologische Nachsorge ist bei Diagnose von MEN2B erforderlich?
A
Bei MEN2B werden regelmäßige Spaltlampenuntersuchungen durchgeführt, um Veränderungen der Hornhautnerven sowie der konjunktivalen und lidständigen Neurome zu beobachten. Überprüfen Sie auf Symptome trockener Augen und behandeln Sie diese gegebenenfalls mit künstlichen Tränen. Aufgrund des Risikos eines Offenwinkelglaukoms werden auch regelmäßige Augeninnendruckmessungen und Gesichtsfelduntersuchungen empfohlen. Zudem sollte bildgebend nach einem Hypophysentumor gesucht werden.
6. Pathophysiologie und detaillierter Pathomechanismus
MEN1 entsteht durch Funktionsverlust eines Tumorsuppressorgens gemäß der Zwei-Treffer-Hypothese. Die erste Mutation (Keimbahnmutation) wird vererbt, und die zweite Mutation (somatische Mutation) tritt erworben hinzu, was zu einem vollständigen Verlust der Menin-Protein-Funktion und einem Verlust der Kontrolle über das Zellwachstum führt.
Menin wird sowohl in endokrinen als auch in nicht-endokrinen Geweben exprimiert, aber der genaue Mechanismus der Karzinogenese ist unbekannt. Hypophysentumoren, die das Chiasma opticum komprimieren, verursachen eine bitemporale Hemianopsie.
Das RET-Protein ist ein Tyrosinkinase-Rezeptor, der an der Differenzierung und Migration sich entwickelnder neuroendokriner Gewebe beteiligt ist. Missense-Mutationen im RET-Gen führen zu einem einzelnen Aminosäureaustausch, der eine konstitutive Aktivierung (Funktionsgewinn) des Proteins bewirkt.
RET wird in mehreren Geweben exprimiert, darunter parafollikuläre Zellen der Schilddrüse (C-Zellen), Nebenschilddrüsen, enterische Nervenganglien, chromaffine Zellen der Nebenniere sowie peripheres und zentrales Nervensystem. Dieses breite Expressionsmuster erklärt die vielfältigen Tumormuster bei MEN2.
Die histopathologische Untersuchung verdickter Hornhautnerven bei MEN2B zeigt nicht myelinisierte Nervenfasern mit Schwann-Zellen. Die Axone haben ein normales Aussehen und ihr Durchmesser variiert zwischen 0,1 und 1,4 nm. Konjunktivale und lidständige Neurome erscheinen als kapsellose Nervenproliferationen.
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. American journal of ophthalmology. 1991;111(3):307-11. doi:10.1016/s0002-9394(14)72314-1. PMID:1672053.
Chang TC, Okafor KC, Cavuoto KM, Dubovy SR, Karp CL. Pediatric Multiple Endocrine Neoplasia Type 2B: Clinicopathological Correlation of Perilimbal Mucosal Neuromas and Treatment of Secondary Open-Angle Glaucoma. Ocular oncology and pathology. 2018;4(3):196-198. doi:10.1159/000484053. PMID:29765955; PMCID:PMC5939668.
de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab. 2015;100(9):3288-3296. doi:10.1210/jc.2015-2015.
Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610. doi:10.1089/thy.2014.0335. PMID:25810047; PMCID:PMC4490627.
Nagaoka R, Sugitani I, Sanada M, Jikuzono T, Okamura R, Igarashi T, et al. The Reality of Multiple Endocrine Neoplasia Type 2B Diagnosis: Awareness of Unique Physical Appearance Is Important. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2018;85(3):178-182. doi:10.1272/jnms.JNMS.2018_85-26. PMID:30135345.
Lam D, Villaret J, Nguyen Kim P, Gabison E, Cochereau I, Doan S. In Vivo Confocal Microscopy of Prominent Conjunctival and Corneal Nerves in Multiple Endocrine Neoplasia Type 2B. Cornea. 2019;38(11):1453-1455. doi:10.1097/ICO.0000000000002028. PMID:31205161.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.