MEN의 안과적 특징은 고전적으로 MEN2B형에서 기술됩니다. MEN2B형에서는 갑상선 수질암, 갈색세포종 외에 점막 신경종과 장관 신경절세포종증이 동반됩니다. 점막 신경종은 MEN2B 환자의 거의 100%에서 발견되며, 이 증후군의 병리학적 특징으로 알려져 있습니다. 안과적 소견은 MEN2B형의 조기 진단에 단서가 될 수 있습니다 5).
MEN1형에서는 Dutch MEN1 연구의 대규모 코호트 (323예)에서 환자의 38.1%에서 뇌하수체 종양이 발생하는 것으로 나타났습니다 3). 프로락틴종이 가장 흔한 뇌하수체 종양이며, 도파민 작용제에 의한 약물 요법에 잘 반응합니다.
QMEN2B형의 안과적 소견은 언제부터 나타납니까?
A
MEN2B형의 특징적인 소견은 소아기부터 인식 가능합니다. 각막 신경 비후와 점막 신경종은 조기에 나타나며, MEN2B형의 첫 번째 임상적 단서가 될 수 있습니다. MEN2B형의 MTC는 유아기에 발병할 수도 있으며, 안과적 소견의 조기 인식이 진단과 예방적 치료 시기를 결정합니다.
현저한 각막 신경은 MEN2B형에서 가장 특징적인 안과적 소견이다. 세극등 현미경 검사로 각막 내 비후된 신경 섬유를 확인한다. 조직병리학적으로는 슈반 세포를 동반한 무수신경이며, 축삭은 정상적인 외관을 보인다. MEN2A형에서도 Kinoshita 등의 보고에 따르면 28안 중 16안(57%)에서 grade 2 이상의 각막 신경 비후를 보였고, 약 29%에서 grade 3~4의 현저한 비후가 관찰되어, MEN 전반에서 중요한 안과적 소견이다1).
결막 및 눈꺼풀의 신경종은 피막이 없는 비후된 신경 증식으로 인지된다. 입술, 혀, 볼 점막에도 유사한 점막 신경종이 나타난다. 생체 공초점 현미경(IVCM)에서는 결막 신경종으로 크고 두꺼운 신경 다발의 불규칙한 배열, 고리, 분지, 최대 1mm에 이르는 확장상이 관찰되며, 각막에서는 기저하 신경총의 비후상이 관찰된다6). 소아 증례에서는 윤부 주위의 점막 신경종이 이차성 개방각 녹내장을 유발하여 안압 관리가 필요하다고 보고되었다2).
MEN1형에서는 뇌하수체 종양에 의한 시교차 증후군이 주요 안과적 문제이다. 시야 검사에서 수직 자오선을 경계로 하는 양측 반맹이 가장 흔하게 관찰된다. 시신경 위축을 동반하기도 한다.
**비안과적 특징(MEN2B형)**으로 입술 비대, 마르판 증후군 유사 체형, 장관 신경절세포종증(거대결장을 유발할 수 있음)이 나타난다.
Q각막 신경이 두꺼워 보이는 경우, 반드시 MEN을 의심해야 합니까?
A
각막 신경의 현저화는 MEN2B형에 특이적인 소견이 아니다. 레프섬 증후군, 한센병, 라일리-데이 증후군, 신경섬유종증, 선천성 어린선에서도 관찰된다. 또한 원추각막, 단순포진 각막염, 후부 다형성 각막 변성 등 각막 질환에서도 각막 신경이 두드러질 수 있다. 그러나 MEN2B형만큼 현저하지는 않다. 다른 전신 소견(입술 비대, 마르판 유사 체형 등)과의 조합으로 판단한다.
RET 원종양 유전자(염색체 10q11.2)의 미스센스 돌연변이가 원인이다. RET 단백질은 티로신 키나제 수용체이며, 신경내분비 조직의 분화와 이동에 관여한다. 돌연변이에 의해 기능 획득형 이상이 발생하여 종양이 발생한다.
MEN2B형에서는 약 95%의 환자에서 코돈 918(M918T 돌연변이)의 생식세포 계열 돌연변이가 확인됩니다4). 이 돌연변이를 가진 환자는 유아기부터 발병하며 가장 침습적인 MTC를 나타냅니다. 한편, MEN2B 환자의 진단은 지연되는 경향이 있으며, Nagaoka 등의 일본 증례 보고에 따르면 4예 모두 유아기부터 점막 신경종 및 위장관 증상이 있었음에도 불구하고 증상성 MTC 단계에서 처음 진단되었습니다5).
Mehtab Ahmad, Imran Rizvi, Amit Jain, Noorin Zaidi Painful Hip Leading to the Diagnosis of MEN 2B Syndrome 2012 Nov 26 Case Rep Endocrinol. 2012 Nov 26; 2012:567060 Figure 2. PMCID: PMC3513729. License: CC BY.
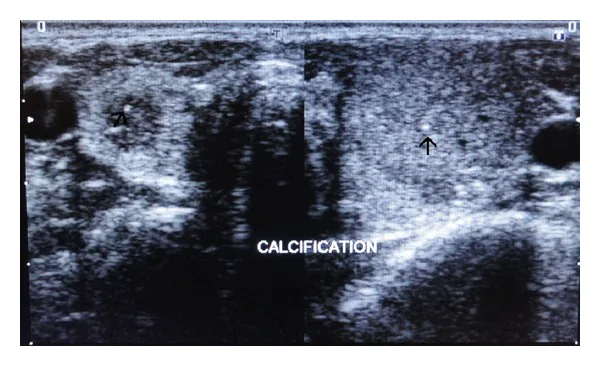
좌우 갑상선 초음파 영상에서 실질 내에 점상 고에코 석회화가 화살표로 표시되어 있습니다. 다발성 내분비 종양에서 문제가 되는 갑상선 병변의 평가 영상입니다.
MEN 증후군의 진단은 일반적으로 가족력과 비안과적 소견에 기반하여 이루어집니다. 안과 검사는 특히 새로운 증례의 정밀 검사에 유용합니다.
안과 의사는 MEN의 안과적 발현을 조기에 발견하고 적절히 의뢰하며 시각 재활에 중요한 역할을 합니다. MEN1형의 뇌하수체 종양에서는 정기적인 시야 검사와 시력 평가가 필요합니다. MEN2B형에서는 안구건조증 관리와 정기적인 안압 측정을 시행합니다.
QMEN2B형으로 진단된 경우, 안과적으로 어떤 경과 관찰이 필요한가요?
A
MEN2B형에서는 정기적인 세극등 검사로 각막 신경과 결막/안검 신경종의 변화를 관찰합니다. 안구건조증 증상 유무를 확인하고 필요에 따라 인공눈물 등으로 관리합니다. 개방각 녹내장의 위험이 있으므로 정기적인 안압 측정과 시야 검사도 권장됩니다. 또한 뇌하수체 종양의 합병 여부를 영상 검사로 평가합니다.
MEN1형은 ‘2회 타격 가설’에 기반한 종양 억제 유전자의 기능 상실로 발병합니다. 첫 번째 돌연변이(생식세포계열 돌연변이)는 유전을 통해 전달되고, 두 번째 돌연변이(체세포 돌연변이)가 후천적으로 추가되어 메닌 단백질의 기능이 완전히 상실되고 세포 증식 조절이 상실됩니다.
메닌은 내분비 조직과 비내분비 조직 모두에서 발현되지만, 발암으로 이어지는 정확한 기전은 아직 밝혀지지 않았습니다. 뇌하수체 종양이 시교차를 압박하여 양측 반맹을 유발합니다.
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. American journal of ophthalmology. 1991;111(3):307-11. doi:10.1016/s0002-9394(14)72314-1. PMID:1672053.
Chang TC, Okafor KC, Cavuoto KM, Dubovy SR, Karp CL. Pediatric Multiple Endocrine Neoplasia Type 2B: Clinicopathological Correlation of Perilimbal Mucosal Neuromas and Treatment of Secondary Open-Angle Glaucoma. Ocular oncology and pathology. 2018;4(3):196-198. doi:10.1159/000484053. PMID:29765955; PMCID:PMC5939668.
de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab. 2015;100(9):3288-3296. doi:10.1210/jc.2015-2015.
Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610. doi:10.1089/thy.2014.0335. PMID:25810047; PMCID:PMC4490627.
Nagaoka R, Sugitani I, Sanada M, Jikuzono T, Okamura R, Igarashi T, et al. The Reality of Multiple Endocrine Neoplasia Type 2B Diagnosis: Awareness of Unique Physical Appearance Is Important. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2018;85(3):178-182. doi:10.1272/jnms.JNMS.2018_85-26. PMID:30135345.
Lam D, Villaret J, Nguyen Kim P, Gabison E, Cochereau I, Doan S. In Vivo Confocal Microscopy of Prominent Conjunctival and Corneal Nerves in Multiple Endocrine Neoplasia Type 2B. Cornea. 2019;38(11):1453-1455. doi:10.1097/ICO.0000000000002028. PMID:31205161.