La neoplasia endocrina múltiple (MEN) es un grupo poco común de trastornos hereditarios con un mayor riesgo de desarrollar neoplasias en dos o más glándulas endocrinas. Sigue un patrón de herencia autosómico dominante con alta penetrancia y expresividad variable.
El MEN se clasifica principalmente en cuatro tipos.
MEN tipo 1
Gen responsable: gen MEN1 (11q13)
Combinación de tumores: tumores paratiroideos, tumores hipofisarios, tumores gastroenteropancreáticos
Prevalencia: 2 a 20 por cada 100,000 personas
Características oftálmicas: Compresión del quiasma óptico por tumor hipofisario (hemianopsia bitemporal)
MEN tipo 2 (2A y 2B)
Gen responsable: Protooncogén RET (10q11.2)
Combinación de tumores: Carcinoma medular de tiroides (CMT), feocromocitoma, hiperparatiroidismo (solo tipo 2A)
Prevalencia: 1 de cada 35,000 personas (para todo MEN tipo 2)
Características oftálmicas: Engrosamiento marcado de los nervios corneales, neuromas conjuntivales y palpebrales (especialmente en MEN tipo 2B)
Las características oftálmicas del MEN se describen clásicamente en el MEN tipo 2B. En el MEN tipo 2B, además del carcinoma medular de tiroides y el feocromocitoma, se presentan neuromas mucosos y ganglioneuromatosis intestinal. Los neuromas mucosos se encuentran en casi el 100% de los pacientes con MEN tipo 2B y se conocen como un hallazgo patognomónico de este síndrome. Los hallazgos oftálmicos pueden proporcionar pistas para el diagnóstico temprano del MEN tipo 2B 5).
En el MEN tipo 1, un gran cohorte del Estudio Holandés MEN1 (323 casos) mostró que el 38.1% de los pacientes desarrollaron tumores hipofisarios 3). El prolactinoma es el tumor hipofisario más común y responde bien al tratamiento farmacológico con agonistas dopaminérgicos.
Q¿A qué edad se reconocen los hallazgos oftálmicos del MEN tipo 2B?
A
Los hallazgos característicos del MEN tipo 2B pueden reconocerse desde la infancia. El engrosamiento de los nervios corneales y los neuromas mucosos aparecen temprano y pueden ser las primeras pistas clínicas del MEN tipo 2B. El CMT en el MEN tipo 2B puede desarrollarse en la lactancia, y el reconocimiento temprano de los hallazgos oftálmicos influye en el momento del diagnóstico y el tratamiento preventivo.
Los nervios corneales prominentes son el hallazgo ocular más característico en el MEN2B. Se observan fibras nerviosas engrosadas en la córnea mediante microscopía con lámpara de hendidura. Histopatológicamente, son nervios amielínicos con células de Schwann, y los axones tienen apariencia normal. En el MEN2A, según el informe de Kinoshita et al., se observó engrosamiento del nervio corneal de grado 2 o superior en 16 de 28 ojos (57%), y engrosamiento marcado de grado 3-4 en aproximadamente el 29%, lo que lo convierte en un hallazgo ocular importante en el MEN en general 1).
Los neuromas conjuntivales y palpebrales se reconocen como proliferaciones nerviosas engrosadas sin cápsula. Neuromas mucosos similares también aparecen en los labios, la lengua y la mucosa bucal. Con microscopía confocal in vivo (IVCM), se observa una disposición irregular de haces nerviosos grandes y gruesos, bucles, ramificaciones y dilataciones de hasta 1 mm en los neuromas conjuntivales, y engrosamiento del plexo nervioso subbasal en la córnea6). En casos pediátricos, se ha informado que los neuromas mucosos alrededor del limbo causan glaucoma secundario de ángulo abierto que requiere manejo de la presión intraocular2).
En el MEN1, el principal problema oftalmológico es el síndrome quiasmático debido a tumores hipofisarios. La hemianopsia bitemporal con límite en el meridiano vertical es la más frecuente en la prueba de campo visual. También puede haber atrofia óptica.
Las características no oftálmicas (MEN2B) incluyen hipertrofia de labios, hábito marfanoide y ganglioneuromatosis intestinal (que puede causar megacolon).
QSi los nervios corneales se ven engrosados, ¿siempre se debe sospechar MEN?
A
La prominencia de los nervios corneales no es específica del MEN2B. También se observa en el síndrome de Refsum, la enfermedad de Hansen, el síndrome de Riley-Day, la neurofibromatosis y la ictiosis congénita. Además, los nervios corneales pueden ser prominentes en enfermedades corneales como el queratocono, la queratitis herpética simple y la distrofia corneal polimorfa posterior. Sin embargo, generalmente no son tan prominentes como en el MEN2B. El diagnóstico se realiza en combinación con otros hallazgos sistémicos (hipertrofia de labios, hábito marfanoide, etc.).
Los síndromes MEN siguen un patrón de herencia autosómico dominante. Los individuos con mutaciones en RET tienen un 50% de riesgo de transmitir la mutación a la descendencia. También existen casos esporádicos debidos a mutaciones somáticas.
Las mutaciones en el gen MEN1 (cromosoma 11q13) son la causa. El gen MEN1 codifica una proteína llamada menina, un supresor tumoral. La pérdida de función ocurre mediante un mecanismo de “dos golpes” con mutaciones en ambos alelos, lo que lleva a una proliferación celular descontrolada.
Las mutaciones de sentido erróneo en el protooncogén RET (cromosoma 10q11.2) son la causa. La proteína RET es un receptor de tirosina quinasa involucrado en la diferenciación y migración de tejidos neuroendocrinos. Las mutaciones resultan en anomalías de ganancia de función, lo que lleva al desarrollo de tumores.
En el MEN2B, aproximadamente el 95% de los pacientes presentan una mutación germinal en el codón 918 (mutación M918T) 4). Los pacientes con esta mutación desarrollan la enfermedad en la infancia y presentan la forma más agresiva de MTC. Por otro lado, el diagnóstico de los pacientes con MEN2B tiende a retrasarse; en una serie de casos japonesa de Nagaoka et al., los cuatro pacientes presentaban neuromas mucosos y síntomas gastrointestinales desde la infancia, pero fueron diagnosticados por primera vez en la etapa de MTC sintomática 5).
Mehtab Ahmad, Imran Rizvi, Amit Jain, Noorin Zaidi Painful Hip Leading to the Diagnosis of MEN 2B Syndrome 2012 Nov 26 Case Rep Endocrinol. 2012 Nov 26; 2012:567060 Figure 2. PMCID: PMC3513729. License: CC BY.
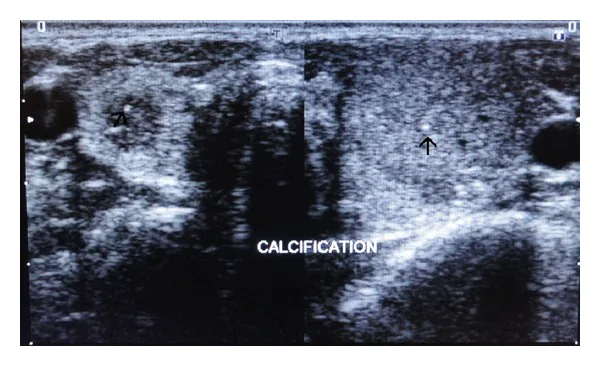
Las ecografías de los tiroides izquierdo y derecho muestran calcificaciones hiperecogénicas puntiformes dentro del parénquima, indicadas con flechas. Esta es una imagen de evaluación de las lesiones tiroideas problemáticas en la neoplasia endocrina múltiple.
El diagnóstico de los síndromes MEN generalmente se basa en la historia familiar y los hallazgos no oftálmicos. El examen oftálmico es particularmente útil para la evaluación detallada en casos nuevos.
Microscopía con lámpara de hendidura: Esencial para observar el engrosamiento de los nervios corneales y los neuromas conjuntivales/palpebrales.
Microscopía confocal in vivo (IVCM): Visualiza de forma no invasiva los característicos haces nerviosos grandes y las estructuras en asa de los neuromas conjuntivales, así como el engrosamiento del plexo nervioso subbasal corneal 6).
Perimetría de Goldmann (GVF): Se utiliza para evaluar el síndrome quiasmático debido a tumores hipofisarios en MEN1. La hemianopsia bitemporal es el hallazgo más frecuente.
Prueba de visión cromática de Ishihara: Se utiliza para evaluar la alteración de la visión cromática debida a tumores hipofisarios.
Tomografía de coherencia óptica de alta resolución (HR-OCT): Se utiliza para la evaluación no invasiva de neuromas conjuntivales y es útil para el diagnóstico diferencial con otras enfermedades.
El consejo genético debe realizarse antes de la prueba genética de RET. En familias con MEN2, el cribado debe realizarse al nacer o lo antes posible. Se debe ofrecer consejo genético a todos los familiares de primer grado de los casos índice recién diagnosticados con mutaciones en RET.
Para pacientes con mutaciones germinales en los codones 883, 918 o 922 en MEN2B, se debe realizar tiroidectomía total y disección de ganglios linfáticos centrales dentro de los primeros meses de vida, debido a que la penetrancia del CMT a lo largo de la vida es casi del 100%.
Los oftalmólogos desempeñan un papel clave en la detección temprana de las manifestaciones oculares en MEN, la derivación adecuada y la rehabilitación visual. Para los tumores hipofisarios en MEN1, son necesarias pruebas periódicas del campo visual y evaluación de la agudeza visual. En MEN2B, se realiza el manejo del ojo seco y la medición periódica de la presión intraocular.
Q¿Qué seguimiento oftalmológico es necesario cuando se diagnostica MEN2B?
A
En MEN2B, se realiza un examen periódico con lámpara de hendidura para observar cambios en los nervios corneales y neuromas conjuntivales/palpebrales. Se deben evaluar los síntomas de ojo seco y manejarlos con lágrimas artificiales según sea necesario. Debido al riesgo de glaucoma de ángulo abierto, también se recomiendan la medición periódica de la presión intraocular y las pruebas del campo visual. Además, se deben realizar estudios de imagen para evaluar la presencia de tumores hipofisarios.
El MEN1 se desarrolla debido a la pérdida de función de un gen supresor de tumores basada en la “hipótesis de dos impactos”. La primera mutación (mutación de la línea germinal) se hereda, y la segunda mutación (mutación somática) se adquiere posteriormente, lo que lleva a la pérdida completa de la función de la proteína menina y a la pérdida del control de la proliferación celular.
La menina se expresa tanto en tejidos endocrinos como no endocrinos, pero el mecanismo exacto que conduce a la carcinogénesis sigue sin aclararse. Los tumores hipofisarios que comprimen el quiasma óptico causan hemianopsia bitemporal.
La proteína RET es un receptor de tirosina quinasa involucrado en la diferenciación y migración de los tejidos neuroendocrinos en desarrollo. Las mutaciones sin sentido en el gen RET resultan en una sustitución de un solo aminoácido, causando una activación constitutiva (ganancia de función) de la proteína.
RET se expresa en múltiples tejidos, incluyendo células parafoliculares tiroideas (células C), glándulas paratiroides, ganglios intestinales, células cromafines suprarrenales y sistemas nerviosos periférico y central. Este amplio patrón de expresión explica los diversos patrones tumorales en el MEN2.
El examen histopatológico de los nervios corneales engrosados en el MEN2B muestra nervios amielínicos con células de Schwann. Los axones tienen una apariencia normal y varían en diámetro entre 0.1 y 1.4 nm. Los neuromas conjuntivales y palpebrales se observan como proliferaciones nerviosas no encapsuladas.
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. American journal of ophthalmology. 1991;111(3):307-11. doi:10.1016/s0002-9394(14)72314-1. PMID:1672053.
Chang TC, Okafor KC, Cavuoto KM, Dubovy SR, Karp CL. Pediatric Multiple Endocrine Neoplasia Type 2B: Clinicopathological Correlation of Perilimbal Mucosal Neuromas and Treatment of Secondary Open-Angle Glaucoma. Ocular oncology and pathology. 2018;4(3):196-198. doi:10.1159/000484053. PMID:29765955; PMCID:PMC5939668.
de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab. 2015;100(9):3288-3296. doi:10.1210/jc.2015-2015.
Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610. doi:10.1089/thy.2014.0335. PMID:25810047; PMCID:PMC4490627.
Nagaoka R, Sugitani I, Sanada M, Jikuzono T, Okamura R, Igarashi T, et al. The Reality of Multiple Endocrine Neoplasia Type 2B Diagnosis: Awareness of Unique Physical Appearance Is Important. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2018;85(3):178-182. doi:10.1272/jnms.JNMS.2018_85-26. PMID:30135345.
Lam D, Villaret J, Nguyen Kim P, Gabison E, Cochereau I, Doan S. In Vivo Confocal Microscopy of Prominent Conjunctival and Corneal Nerves in Multiple Endocrine Neoplasia Type 2B. Cornea. 2019;38(11):1453-1455. doi:10.1097/ICO.0000000000002028. PMID:31205161.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.