La neoplasia endocrina multipla (MEN) è un raro gruppo di malattie ereditarie che aumentano il rischio di sviluppare neoplasie in due o più ghiandole endocrine. Segue una modalità di trasmissione autosomica dominante, con elevata penetranza ed espressività variabile.
La MEN è classificata principalmente in quattro tipi.
Prevalenza : da 2 a 20 persone ogni 100.000 abitanti
Caratteristiche oftalmologiche: compressione chiasmatica da tumore ipofisario (emianopsia bitemporale)
MEN tipo 2 (2A e 2B)
Gene responsabile: proto-oncogene RET (10q11.2)
Associazione tumorale: carcinoma midollare della tiroide (CMT), feocromocitoma, iperparatiroidismo (solo nel tipo 2A)
Prevalenza: 1 persona su 35.000 (per l’intero MEN tipo 2)
Caratteristiche oftalmologiche: marcato ispessimento dei nervi corneali, neuromi congiuntivali e palpebrali (soprattutto nel MEN tipo 2B)
Le caratteristiche oftalmologiche del MEN sono classicamente descritte nel MEN tipo 2B. Il MEN tipo 2B associa carcinoma midollare della tiroide, feocromocitoma, neuromi mucosi e ganglioneuromatosi intestinale. I neuromi mucosi sono presenti in quasi il 100% dei pazienti con MEN2B e sono considerati un segno patognomonico di questa sindrome. I reperti oftalmologici possono essere un indizio per la diagnosi precoce del MEN2B 5).
Nel MEN tipo 1, lo studio DutchMEN1 su una grande coorte (323 casi) ha mostrato che il 38,1% dei pazienti sviluppa un tumore ipofisario 3). Il prolattinoma è il tumore ipofisario più comune e risponde bene alla terapia farmacologica con agonisti dopaminergici.
QA che età compaiono i reperti oftalmologici del MEN2B?
A
I segni caratteristici del MEN2B sono riconoscibili già nell’infanzia. L’ispessimento dei nervi corneali e i neuromi mucosi compaiono precocemente e possono essere il primo indizio clinico del MEN2B. Il CMT nel MEN2B può insorgere nell’infanzia, e il riconoscimento precoce dei reperti oftalmologici influenza i tempi della diagnosi e del trattamento preventivo.
Nervi corneali prominenti sono il reperto oftalmologico più caratteristico del MEN2B. L’esame con lampada a fessura mostra fibre nervose ispessite nella cornea. Istopatologicamente si tratta di nervi amielinici con cellule di Schwann, e gli assoni hanno un aspetto normale. Anche nel MEN2A, secondo il rapporto di Kinoshita et al., in 16 occhi su 28 (57%) è stato riscontrato un ispessimento dei nervi corneali di grado 2 o superiore, e in circa il 29% è stato osservato un marcato ispessimento di grado 3-4, costituendo un importante reperto oftalmologico in generale per il MEN 1).
Neuromi congiuntivali e palpebrali si presentano come proliferazioni nervose ispessite senza capsula. Neuromi mucosi simili compaiono anche su labbra, lingua e mucosa orale. Alla microscopia confocale in vivo (IVCM), nei neuromi congiuntivali si osservano grandi fasci nervosi spessi con disposizione irregolare, anse, ramificazioni e dilatazioni fino a 1 mm, mentre nella cornea si osserva un ispessimento del plesso nervoso subbasale 6). Nei bambini, è stato riportato che i neuromi mucosi perilimbici possono causare glaucoma secondario ad angolo aperto, richiedendo la gestione della pressione intraoculare2).
Nel MEN1, il principale problema oftalmologico è la sindrome chiasmatica dovuta a tumore ipofisario. All’esame del campo visivo, l’emianopsia bitemporale rispettante la linea mediana verticale è la più frequente. Può essere associata ad atrofia ottica.
Caratteristiche non oftalmologiche (MEN2B) includono ipertrofia delle labbra, habitus marfanoide e ganglioneuromatosi intestinale (che può causare megacolon).
QSe i nervi corneali appaiono spessi, si deve sempre sospettare un MEN?
A
La prominenza dei nervi corneali non è specifica del MEN2B. Si riscontra anche nella sindrome di Refsum, nella lebbra, nella sindrome di Riley-Day, nella neurofibromatosi e nell’ittiosi congenita. Inoltre, in malattie corneali come il cheratocono, la cheratite erpetica semplice e la distrofia corneale polimorfa posteriore, i nervi corneali possono essere evidenti. Tuttavia, di solito non sono così marcati come nel MEN2B. La diagnosi si basa sulla combinazione con altri segni sistemici (ipertrofia delle labbra, habitus marfanoide, ecc.).
La sindrome MEN segue una trasmissione autosomica dominante. Gli individui con una mutazione RET hanno un rischio di trasmissione ai discendenti del 50%. Esistono anche casi sporadici dovuti a mutazioni somatiche.
La causa è una mutazione del gene MEN1 (cromosoma 11q13). Il gene MEN1 codifica per la menina, una proteina oncosoppressore. La perdita di funzione di entrambi gli alleli (doppio colpo) porta a una proliferazione cellulare incontrollata.
La causa è una mutazione missenso del proto-oncogene RET (cromosoma 10q11.2). La proteina RET è un recettore tirosin-chinasico coinvolto nella differenziazione e migrazione dei tessuti neuroendocrini. La mutazione porta a un’anomala iperfunzione, con conseguente sviluppo di tumori.
Nel tipo MEN2B, circa il 95% dei pazienti presenta una mutazione germinale al codone 918 (mutazione M918T) 4). I pazienti con questa mutazione sviluppano la malattia nell’infanzia e presentano il MTC più aggressivo. D’altra parte, la diagnosi dei pazienti MEN2B tende ad essere ritardata; in una serie di casi giapponesi riportata da Nagaoka et al., tutti e 4 i casi presentavano neuromi mucosi e sintomi gastrointestinali fin dall’infanzia, ma sono stati diagnosticati solo allo stadio di MTC sintomatico 5).
Mehtab Ahmad, Imran Rizvi, Amit Jain, Noorin Zaidi Painful Hip Leading to the Diagnosis of MEN 2B Syndrome 2012 Nov 26 Case Rep Endocrinol. 2012 Nov 26; 2012:567060 Figure 2. PMCID: PMC3513729. License: CC BY.
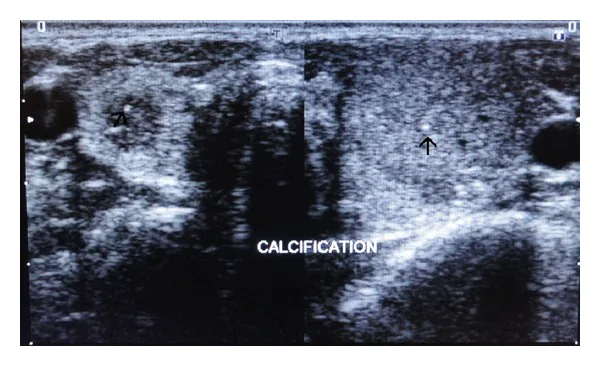
Immagini ecografiche dei lobi tiroidei destro e sinistro che mostrano calcificazioni puntiformi iperecogene nel parenchima (indicate dalle frecce). Si tratta di un’immagine di valutazione delle lesioni tiroidee problematiche nella neoplasia endocrina multipla.
La diagnosi della sindrome MEN si basa solitamente sull’anamnesi familiare e sui reperti non oculistici. L’esame oculistico è particolarmente utile nella valutazione dei nuovi casi.
Esame con lampada a fessura: essenziale per osservare l’ispessimento dei nervi corneali, i neuromi congiuntivali e palpebrali
Microscopia confocale in vivo (IVCM): consente di visualizzare in modo non invasivo i caratteristici fasci nervosi grandi e le strutture ad ansa dei neuromi congiuntivali, nonché l’ispessimento del plesso nervoso subbasale corneale 6)
Perimetro di Goldmann (GVF): utilizzato per valutare la sindrome chiasmatica dovuta a tumore ipofisario nel tipo MEN1. L’emianopsia bitemporale è il reperto più frequente.
Test di visione dei colori di Ishihara: utilizzato per valutare i disturbi della visione dei colori dovuti a tumore ipofisario
Tomografia a coerenza ottica ad alta risoluzione (HR-OCT): utilizzata per la valutazione non invasiva dei neuromi congiuntivali, utile per la diagnosi differenziale con altre malattie
La consulenza genetica deve precedere il test genetico del gene RET. Nelle famiglie con MEN2, lo screening deve essere effettuato alla nascita o il più precocemente possibile. A tutti i parenti di primo grado dei casi indice con nuova diagnosi di mutazione RET dovrebbe essere offerta una consulenza genetica.
Nei pazienti con MEN2B e mutazioni germinali ai codoni 883, 918 o 922, entro i primi mesi di vita deve essere eseguita una tiroidectomia totale con dissezione linfonodale centrale, a causa della penetranza quasi del 100% del MTC nell’arco della vita.
L’oftalmologo svolge un ruolo chiave nella diagnosi precoce delle manifestazioni oftalmiche nella MEN, nell’invio appropriato e nella riabilitazione visiva. Per i tumori ipofisari nella MEN1 sono necessari esami regolari del campo visivo e valutazione dell’acuità visiva. Nella MEN2B si effettuano la gestione dell’occhio secco e la misurazione regolare della pressione intraoculare.
QIn caso di diagnosi di MEN2B, quale follow-up oftalmologico è necessario?
A
Nella MEN2B si eseguono regolari esami con lampada a fessura per osservare i cambiamenti dei nervi corneali e dei neuromi congiuntivali e palpebrali. Verificare la presenza di sintomi di occhio secco e gestirli con lacrime artificiali se necessario. A causa del rischio di glaucoma ad angolo aperto, si raccomandano anche misurazioni regolari della pressione intraoculare ed esami del campo visivo. Inoltre, valutare con imaging la presenza di un tumore ipofisario.
La MEN1 insorge per perdita di funzione di un gene oncosoppressore secondo l’ipotesi dei due colpi. La prima mutazione (germinale) è ereditaria, e la seconda mutazione (somatica) si aggiunge successivamente, causando la completa perdita di funzione della proteina menina e la perdita del controllo della proliferazione cellulare.
La menina è espressa sia nei tessuti endocrini che non endocrini, ma il meccanismo esatto che porta alla cancerogenesi è sconosciuto. I tumori ipofisari che comprimono il chiasma ottico causano emianopsia bitemporale.
La proteina RET è un recettore tirosin-chinasico coinvolto nella differenziazione e migrazione dei tessuti neuroendocrini in via di sviluppo. Le mutazioni missenso del gene RET portano alla sostituzione di un singolo amminoacido, causando l’attivazione costitutiva (guadagno di funzione) della proteina.
RET è espresso in diversi tessuti, tra cui le cellule parafollicolari della tiroide (cellule C), le paratiroidi, i gangli nervosi intestinali, le cellule cromaffini surrenaliche e i sistemi nervosi periferico e centrale. Questo ampio pattern di espressione spiega la varietà dei tumori nella MEN2.
L’esame istopatologico dei nervi corneali ispessiti nella MEN2B mostra fibre nervose amieliniche accompagnate da cellule di Schwann. Gli assoni hanno un aspetto normale e il loro diametro varia tra 0,1 e 1,4 nm. I neuromi congiuntivali e palpebrali appaiono come proliferazioni nervose senza capsula.
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. American journal of ophthalmology. 1991;111(3):307-11. doi:10.1016/s0002-9394(14)72314-1. PMID:1672053.
Chang TC, Okafor KC, Cavuoto KM, Dubovy SR, Karp CL. Pediatric Multiple Endocrine Neoplasia Type 2B: Clinicopathological Correlation of Perilimbal Mucosal Neuromas and Treatment of Secondary Open-Angle Glaucoma. Ocular oncology and pathology. 2018;4(3):196-198. doi:10.1159/000484053. PMID:29765955; PMCID:PMC5939668.
de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab. 2015;100(9):3288-3296. doi:10.1210/jc.2015-2015.
Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610. doi:10.1089/thy.2014.0335. PMID:25810047; PMCID:PMC4490627.
Nagaoka R, Sugitani I, Sanada M, Jikuzono T, Okamura R, Igarashi T, et al. The Reality of Multiple Endocrine Neoplasia Type 2B Diagnosis: Awareness of Unique Physical Appearance Is Important. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2018;85(3):178-182. doi:10.1272/jnms.JNMS.2018_85-26. PMID:30135345.
Lam D, Villaret J, Nguyen Kim P, Gabison E, Cochereau I, Doan S. In Vivo Confocal Microscopy of Prominent Conjunctival and Corneal Nerves in Multiple Endocrine Neoplasia Type 2B. Cornea. 2019;38(11):1453-1455. doi:10.1097/ICO.0000000000002028. PMID:31205161.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.