La néoplasie endocrinienne multiple (NEM) est un groupe rare de maladies héréditaires qui augmentent le risque de développer des néoplasies dans au moins deux glandes endocrines. Elle suit un mode de transmission autosomique dominant, avec une pénétrance élevée et une expressivité variable.
La NEM est principalement classée en quatre types.
Prévalence : 2 à 20 personnes pour 100 000 habitants
Caractéristiques ophtalmologiques : compression chiasmatique par tumeur hypophysaire (hémianopsie bitemporale)
MEN de type 2 (2A et 2B)
Gène responsable : proto-oncogène RET (10q11.2)
Association tumorale : carcinome médullaire de la thyroïde (CMT), phéochromocytome, hyperparathyroïdie (type 2A uniquement)
Prévalence : 1 personne sur 35 000 (pour l’ensemble du MEN de type 2)
Caractéristiques ophtalmologiques : épaississement marqué des nerfs cornéens, névromes conjonctivaux et palpébraux (surtout dans le MEN de type 2B)
Les caractéristiques ophtalmologiques du MEN sont classiquement décrites dans le MEN de type 2B. Le MEN de type 2B associe un carcinome médullaire de la thyroïde, un phéochromocytome, des névromes muqueux et une ganglioneuromatose intestinale. Les névromes muqueux sont présents chez près de 100 % des patients atteints de MEN2B et sont considérés comme une caractéristique pathognomonique de ce syndrome. Les signes ophtalmologiques peuvent être une clé pour le diagnostic précoce du MEN2B 5).
Dans le MEN de type 1, l’étude DutchMEN1 sur une large cohorte (323 cas) a montré que 38,1 % des patients développent une tumeur hypophysaire 3). Le prolactinome est la tumeur hypophysaire la plus fréquente et répond bien au traitement médicamenteux par agonistes dopaminergiques.
QÀ quel âge les signes ophtalmologiques du MEN2B apparaissent-ils ?
A
Les signes caractéristiques du MEN2B sont reconnaissables dès l’enfance. L’épaississement des nerfs cornéens et les névromes muqueux apparaissent précocement et peuvent constituer le premier indice clinique du MEN2B. Le CMT du MEN2B peut survenir dès la petite enfance, et la reconnaissance précoce des signes ophtalmologiques influence le moment du diagnostic et du traitement préventif.
Les nerfs cornéens proéminents sont la manifestation ophtalmologique la plus caractéristique du MEN2B. L’examen à la lampe à fente révèle des fibres nerveuses épaissies dans la cornée. Histopathologiquement, ce sont des nerfs amyéliniques avec des cellules de Schwann, et les axones ont un aspect normal. Dans le MEN2A, selon le rapport de Kinoshita et al., un épaississement des nerfs cornéens de grade 2 ou plus a été observé dans 16 yeux sur 28 (57 %), et un épaississement marqué de grade 3 à 4 a été observé dans environ 29 % des cas, ce qui constitue un signe ophtalmologique important dans l’ensemble du MEN 1).
Les neuromas conjonctivaux et palpébraux se présentent comme des proliférations nerveuses épaissies sans capsule. Des neuromas muqueux similaires apparaissent également sur les lèvres, la langue et la muqueuse buccale. En microscopie confocale in vivo (IVCM), on observe dans les neuromas conjonctivaux de grands faisceaux nerveux épais disposés de manière irrégulière, avec des boucles, des branches et des dilatations atteignant jusqu’à 1 mm, tandis que dans la cornée, on observe un épaississement du plexus nerveux sous-basal 6). Chez les enfants, il a été rapporté que les neuromas muqueux péri-limbiques peuvent provoquer un glaucome secondaire à angle ouvert, nécessitant une gestion de la pression intraoculaire2).
Dans le MEN1, le principal problème ophtalmologique est le syndrome chiasmatique dû à une tumeur hypophysaire. L’hémianopsie bitemporale respectant la ligne médiane verticale est la plus fréquente à l’examen du champ visuel. Elle peut être accompagnée d’une atrophie optique.
Les caractéristiques non ophtalmologiques (MEN2B) comprennent une hypertrophie des lèvres, un habitus de type syndrome de Marfan et une ganglioneuromatose intestinale (pouvant provoquer un mégacôlon).
QSi les nerfs cornéens semblent épais, faut-il toujours suspecter un MEN ?
A
La proéminence des nerfs cornéens n’est pas spécifique au MEN2B. On la retrouve également dans le syndrome de Refsum, la lèpre, le syndrome de Riley-Day, la neurofibromatose et l’ichtyose congénitale. De plus, certaines maladies cornéennes comme le kératocône, la kératite herpétique simple et la dystrophie endothéliale polymorphe postérieure peuvent également rendre les nerfs cornéens plus visibles. Cependant, ils ne sont généralement pas aussi marqués que dans le MEN2B. Le diagnostic repose sur la combinaison avec d’autres signes systémiques (hypertrophie des lèvres, habitus marfanoïde, etc.).
Le syndrome MEN est transmis selon un mode autosomique dominant. Les personnes porteuses d’une mutation RET ont un risque de transmission à leur descendance de 50 %. Des cas sporadiques dus à des mutations somatiques existent également.
La cause est une mutation du gène MEN1 (chromosome 11q13). Le gène MEN1 code pour la ménine, une protéine suppresseur de tumeur. La perte de fonction par « double hit » (mutation des deux allèles) entraîne une prolifération cellulaire incontrôlée.
La cause est une mutation faux-sens du proto-oncogène RET (chromosome 10q11.2). La protéine RET est un récepteur à tyrosine kinase impliqué dans la différenciation et la migration des tissus neuroendocriniens. La mutation entraîne un gain de fonction anormal, conduisant au développement de tumeurs.
Dans le type MEN2B, environ 95 % des patients présentent une mutation germinale au codon 918 (mutation M918T) 4). Les patients porteurs de cette mutation développent la maladie dès la petite enfance et présentent le CMT le plus agressif. Cependant, le diagnostic des patients MEN2B a tendance à être retardé ; dans une série de cas japonais rapportée par Nagaoka et al., les 4 cas présentaient des neuromuqueuses et des symptômes gastro-intestinaux dès la petite enfance, mais n’ont été diagnostiqués qu’au stade du CMT symptomatique 5).
Mehtab Ahmad, Imran Rizvi, Amit Jain, Noorin Zaidi Painful Hip Leading to the Diagnosis of MEN 2B Syndrome 2012 Nov 26 Case Rep Endocrinol. 2012 Nov 26; 2012:567060 Figure 2. PMCID: PMC3513729. License: CC BY.
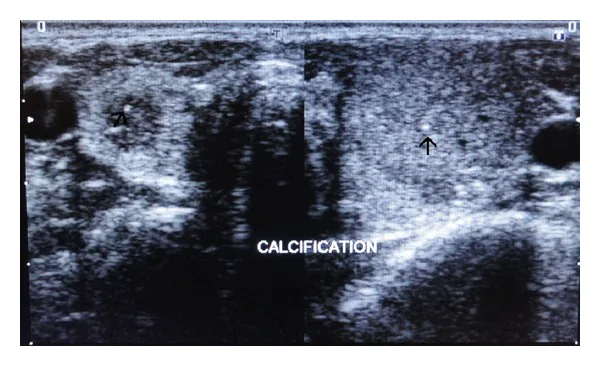
Images échographiques des lobes thyroïdiens droit et gauche montrant des calcifications ponctuées hyperéchogènes dans le parenchyme (indiquées par des flèches). Il s’agit d’une image d’évaluation des lésions thyroïdiennes problématiques dans la néoplasie endocrinienne multiple.
Le diagnostic du syndrome MEN repose généralement sur les antécédents familiaux et les résultats non ophtalmologiques. L’examen ophtalmologique est particulièrement utile pour l’évaluation des nouveaux cas.
Examen à la lampe à fente : essentiel pour observer l’épaississement des nerfs cornéens et les neuromuqueuses conjonctivales et palpébrales
Microscopie confocale in vivo (IVCM) : permet de visualiser de manière non invasive les faisceaux nerveux volumineux caractéristiques et les structures en boucle des neuromuqueuses conjonctivales, ainsi que l’épaississement du plexus nerveux sous-basal cornéen 6)
Périmètre de Goldmann (GVF) : utilisé pour évaluer le syndrome chiasmatique dû à une tumeur hypophysaire dans le type MEN1. L’hémianopsie bitemporale est la constatation la plus fréquente.
Test de vision des couleurs d’Ishihara : utilisé pour évaluer les troubles de la vision des couleurs dus à une tumeur hypophysaire
Tomographie par cohérence optique à haute résolution (HR-OCT) : utilisée pour l’évaluation non invasive des neuromuqueuses conjonctivales, utile pour le diagnostic différentiel avec d’autres maladies
La prise en charge des patients atteints de NEM nécessite une collaboration multidisciplinaire impliquant des généticiens cliniciens, des endocrinologues, des chirurgiens et des oncologues.
Le conseil génétique doit précéder le test génétique du gène RET. Dans les familles atteintes de NEM2, le dépistage doit être effectué à la naissance ou dès que possible. Un conseil génétique doit être proposé à tous les parents au premier degré des cas index nouvellement diagnostiqués avec une mutation RET.
Chez les patients atteints de NEM2B avec des mutations germinales aux codons 883, 918 ou 922, une thyroïdectomie totale avec curage ganglionnaire central doit être réalisée dans les premiers mois de vie, en raison de la pénétrance quasi totale du CMT à vie.
L’ophtalmologiste joue un rôle clé dans la détection précoce des manifestations ophtalmiques de la NEM, l’orientation appropriée et la réadaptation visuelle. Pour les tumeurs hypophysaires dans la NEM1, des examens réguliers du champ visuel et de l’acuité visuelle sont nécessaires. Dans la NEM2B, une gestion de la sécheresse oculaire et une mesure régulière de la pression intraoculaire sont effectuées.
QEn cas de diagnostic de NEM2B, quel suivi ophtalmologique est nécessaire ?
A
Dans la NEM2B, un examen régulier à la lampe à fente permet d’observer les modifications des nerfs cornéens et des névromes conjonctivaux et palpébraux. Vérifier la présence de symptômes de sécheresse oculaire et les gérer avec des larmes artificielles si nécessaire. En raison du risque de glaucome à angle ouvert, une mesure régulière de la pression intraoculaire et un examen du champ visuel sont également recommandés. De plus, évaluer par imagerie la présence éventuelle de tumeur hypophysaire.
La NEM1 résulte d’une perte de fonction du gène suppresseur de tumeur selon l’hypothèse des deux hits. La première mutation (germinale) est héritée, et la seconde mutation (somatique) survient de manière acquise, entraînant une perte complète de la fonction de la protéine ménine et une perte de contrôle de la prolifération cellulaire.
La ménine est exprimée à la fois dans les tissus endocriniens et non endocriniens, mais le mécanisme exact conduisant à la cancérogenèse reste inconnu. Les tumeurs hypophysaires comprimant le chiasma optique provoquent une hémianopsie bitemporale.
La protéine RET est un récepteur tyrosine kinase impliqué dans la différenciation et la migration des tissus neuroendocriniens en développement. Les mutations faux-sens du gène RET entraînent une substitution d’un seul acide aminé, provoquant une activation constitutive (gain de fonction) de la protéine.
RET est exprimé dans plusieurs tissus, notamment les cellules parafolliculaires de la thyroïde (cellules C), les glandes parathyroïdes, les ganglions nerveux intestinaux, les cellules chromaffines surrénaliennes, et les systèmes nerveux périphérique et central. Ce large profil d’expression explique la diversité des tumeurs dans la NEM2.
Histopathologie des manifestations ophtalmologiques
L’examen histopathologique des nerfs cornéens épaissis dans la NEM2B montre des fibres nerveuses non myélinisées accompagnées de cellules de Schwann. Les axones ont une apparence normale et leur diamètre varie de 0,1 à 1,4 nm. Les névromes conjonctivaux et palpébraux apparaissent comme des proliférations nerveuses sans capsule.
Kinoshita S, Tanaka F, Ohashi Y, Ikeda M, Takai S. Incidence of prominent corneal nerves in multiple endocrine neoplasia type 2A. American journal of ophthalmology. 1991;111(3):307-11. doi:10.1016/s0002-9394(14)72314-1. PMID:1672053.
Chang TC, Okafor KC, Cavuoto KM, Dubovy SR, Karp CL. Pediatric Multiple Endocrine Neoplasia Type 2B: Clinicopathological Correlation of Perilimbal Mucosal Neuromas and Treatment of Secondary Open-Angle Glaucoma. Ocular oncology and pathology. 2018;4(3):196-198. doi:10.1159/000484053. PMID:29765955; PMCID:PMC5939668.
de Laat JM, Dekkers OM, Pieterman CRC, Kluijfhout WP, Hermus AR, Pereira AM, et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J Clin Endocrinol Metab. 2015;100(9):3288-3296. doi:10.1210/jc.2015-2015.
Wells SA, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, Lee N, Machens A, Moley JF, Pacini F, Raue F, Frank-Raue K, Robinson B, Rosenthal MS, Santoro M, Schlumberger M, Shah M, Waguespack SG, American Thyroid Association Guidelines Task Force on Medullary Thyroid Carcinoma. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;25(6):567-610. doi:10.1089/thy.2014.0335. PMID:25810047; PMCID:PMC4490627.
Nagaoka R, Sugitani I, Sanada M, Jikuzono T, Okamura R, Igarashi T, et al. The Reality of Multiple Endocrine Neoplasia Type 2B Diagnosis: Awareness of Unique Physical Appearance Is Important. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2018;85(3):178-182. doi:10.1272/jnms.JNMS.2018_85-26. PMID:30135345.
Lam D, Villaret J, Nguyen Kim P, Gabison E, Cochereau I, Doan S. In Vivo Confocal Microscopy of Prominent Conjunctival and Corneal Nerves in Multiple Endocrine Neoplasia Type 2B. Cornea. 2019;38(11):1453-1455. doi:10.1097/ICO.0000000000002028. PMID:31205161.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.