Typ I (Papillenödem-Typ)
Läsion: Gefäße der vorderen Lamina cribrosa
Mechanismus: Venöse Stauung durch unspezifische Entzündung
Merkmale: Vorwiegend Papillenödem. Es gibt Berichte, dass Steroide wirksam sind.

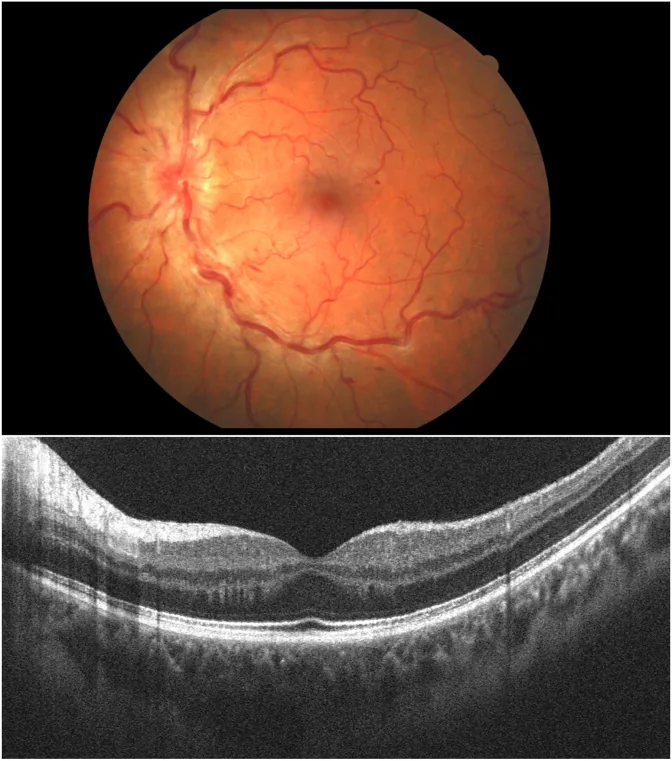
Papillophlebitis (Papillophlebitis) ist eine klinische Subtyp (inkomplette Form) des Zentralvenenverschlusses der Netzhaut (CRVO), eine seltene Erkrankung bei jungen gesunden Erwachsenen unter 45 Jahren1, 2). Sie wird auch als „Optikusdiskussvaskulitis“ bezeichnet.
Während die CRVO bei Menschen mittleren Alters auf Alterung, Bluthochdruck und Arteriosklerose zurückzuführen ist, tritt die Papillophlebitis charakteristischerweise bei gesunden jungen Erwachsenen im Alter von 20–35 Jahren auf, insbesondere bei Frauen. Hayreh (1972) klassifizierte diese Erkrankung in die folgenden zwei Typen.
Typ I (Papillenödem-Typ)
Läsion: Gefäße der vorderen Lamina cribrosa
Mechanismus: Venöse Stauung durch unspezifische Entzündung
Merkmale: Vorwiegend Papillenödem. Es gibt Berichte, dass Steroide wirksam sind.
Typ II (Zentralvenenverschluss-Typ)
Läsion : Sehnervenkopf oder hintere Siebplattenregion
Mechanismus : Entzündung der zentralen Netzhautvene
Merkmale : Venenerweiterung, Schlängelung und Netzhautblutungen. Es gibt Berichte über fehlendes Ansprechen auf Steroide
CRVO tritt häufig bei älteren Menschen mit Risikofaktoren wie Bluthochdruck, Diabetes und Arteriosklerose auf, und der ischämische Typ führt leicht zu schweren Sehstörungen. Papillophlebitis hingegen tritt bei jungen gesunden Menschen auf, das Sehvermögen bleibt oft relativ erhalten, und es handelt sich um eine gutartige Erkrankung, die innerhalb von 6–12 Monaten spontan abklingt. Bei jungen CRVO-Patienten ist die Abgrenzung zur Papillophlebitis jedoch wichtig, und eine systemische Steroidtherapie kann erforderlich sein.
Die Ätiologie wird oft als idiopathisch angenommen, aber es wurde ein Zusammenhang mit Hyperkoagulabilitätszuständen berichtet.
Die folgende Tabelle zeigt die wichtigsten Risikofaktoren und die Häufigkeit systemischer Begleiterkrankungen.
| Risikofaktoren / Begleiterkrankungen | Häufigkeit / Besonderheiten |
|---|---|
| Diabetes | 3–9 % |
| Bluthochdruck | 23–42 % |
| Entzündliche Darmerkrankungen, Psoriasis | Assoziiert |
| Schwangerschaft, orale Kontrazeptiva | Assoziiert |
| Höhenlage, Dehydratation | Assoziiert |
Auch die Assoziation mit hereditären Gerinnungsstörungen ist wichtig. Die Faktor-V-Leiden-Mutation (FVL), die häufigste hereditäre Hyperkoagulabilitätsstörung in Europa, hat in Griechenland eine hohe heterozygote Prävalenz von 15 % (höchste in Europa), und es wurden Fälle von Papillophlebitis aufgrund der Kombination von heterozygoten FVL- und MTHFR-C677T-Mutationen berichtet 2).
Auch der Zusammenhang mit COVID-19 wird beachtet. SARS-CoV-2 verursacht über den ACE2-Rezeptor vaskuläre Endothelschäden und aktiviert das Gerinnungssystem. Bei über 30 % der COVID-19-Patienten treten venöse und arterielle thromboembolische Ereignisse auf, und es wurden Fälle von Papillophlebitis 6 Wochen nach der Infektion berichtet 1).
Sie tritt häufig bei jungen Frauen im Alter von 20–35 Jahren auf. Gerinnungsstörungen (wie Faktor-V-Leiden-Mutation), Zustand nach COVID-19-Erkrankung und die Einnahme oraler Kontrazeptiva werden als Risikofaktoren berichtet. In einigen Fällen liegen auch systemische Erkrankungen (Bluthochdruck, Diabetes) vor.
Die Papillitis venosa ist eine Ausschlussdiagnose, und die Abgrenzung zu häufigen Erkrankungen derselben Altersgruppe wie Stauungspapille und Optikusneuritis ist wichtig.
Zur Bestätigung der Diagnose und Ursachensuche werden folgende Untersuchungen durchgeführt.
Zu den abzugrenzenden Erkrankungen gehören die folgenden.
Derzeit gibt es keine evidenzbasierte, allgemein akzeptierte Behandlung.
Kortikosteroide sind die Hauptbehandlung, die aufgrund einer vermuteten Vaskulitis eingesetzt werden. Bei Typ I wirksam, bei Typ II jedoch Berichten zufolge unwirksam. Da die Erkrankung auch ohne Behandlung spontan ausheilen kann, gibt es keine eindeutigen Belege für die Überlegenheit von Steroiden. Klinisch wird eine systemische Steroidtherapie oft nur bei schweren Fällen in Betracht gezogen.
Ntora et al. (2021) berichteten über eine 22-jährige Frau (heterozygot für FVL und MTHFR-C677T), die mit oralem Methylprednisolon 50 mg/Tag behandelt wurde, mit anschließender Dosisreduktion. Innerhalb einer Woche zeigte sich eine Besserung der Befunde, nach einem Monat eine vollständige Rückbildung. Sehschärfe und Gesichtsfeld blieben nach sechs Monaten normal2).
Als weitere Behandlungen werden auch Thrombozytenaggregationshemmer und Antikoagulanzien vorgeschlagen, deren Wirksamkeit jedoch nicht belegt ist.
Bei schwerem Makulaödem kann eine intravitreale Anti-VEGF-Injektion in Betracht gezogen werden (nicht von der FDA zugelassen).
Insausti-García et al. (2022) behandelten einen 40-jährigen Mann, der nach einer COVID-19-Erkrankung eine Papillitis entwickelte, zunächst mit Aspirin 100 mg/Tag oral und Bromfenac 0,9 mg/mL Augentropfen (2-mal täglich). Aufgrund einer Sehverschlechterung (20/200) durch ein Makulaödem wurde ein intravitreales Dexamethason-Freisetzungsimplantat (Ozurdex) injiziert, und die Sehkraft besserte sich nach zwei Wochen auf 20/401).
Es handelt sich um eine Erkrankung, die sich auch ohne Behandlung spontan bessern kann, und die Prognose ist im Allgemeinen gut, mit einer spontanen Rückbildung innerhalb von 6–12 Monaten. Allerdings besteht bei bis zu 30 % ein Risiko des Fortschreitens zu einem ischämischen Venenverschluss, sodass eine regelmäßige augenärztliche Nachsorge unerlässlich ist. Siehe Abschnitt „Standardbehandlung“ für Details.
Die Pathophysiologie der Papillitis venosa ist unklar, und es wird angenommen, dass der Mechanismus nicht einheitlich ist.
Die Haupthypothese besagt, dass eine idiopathische Entzündung der Sehnervenpapille die zentrale Netzhautvene komprimiert, was zu einer sekundären venösen Insuffizienz und Netzhautblutungen führt. In der Hayreh-Klassifikation umfasst Typ I eine unspezifische Entzündung der Gefäße der Lamina cribrosa anterior, während Typ II eine Entzündung der zentralen Netzhautvene an der Sehnervenpapille oder der Lamina cribrosa posterior umfasst.
COVID-19-bedingte Mechanismen: Die folgende Kaskade wurde vorgeschlagen 1).
Das Muster der COVID-19-assoziierten Gerinnungsstörung (CAC) ist gekennzeichnet durch einen Anstieg von D-Dimer und Fibrinogen parallel zu den Entzündungsmarkern1).
Beteiligung erblicher Gerinnungsstörungen wurde ebenfalls berichtet. Die Kombination von FVL-Heterozygotie und MTHFR-C677T-Heterozygotie kann zu einem Gerinnungsungleichgewicht führen und zur Entstehung einer Papillenphlebitis beitragen2). Die MTHFR-C677T-Mutation kann in homozygoter Form eine Hyperhomocysteinämie verursachen und zu einem hyperkoagulablen Zustand beitragen, aber eine alleinige Heterozygotie führt möglicherweise nicht zu einer Hyperhomocysteinämie.
Der Pathomechanismus ist nicht einheitlich, aber es wird angenommen, dass eine Entzündung der Sehnervenpapille die Zentralvene der Netzhaut komprimiert. Bei jungen Menschen fehlen Risikofaktoren für CRVO wie Bluthochdruck und Arteriosklerose bei Menschen mittleren Alters, sodass Gerinnungsstörungen (erbliche Faktoren) und postinfektiöse Entzündungen (wie COVID-19) relativ wichtige Auslöser darstellen.
Insausti-García et al. (2022) berichteten über einen Fall eines 40-jährigen Mannes, der 6 Wochen nach einer COVID-19-Erkrankung eine Papillophlebitis entwickelte1). Die Untersuchungen zeigten erhöhte Gerinnungs- und Entzündungsmarker: D-Dimer 672 μg/L (normal <460), Fibrinogen 451 mg/dL (normal 200–400), CRP 0,898 mg/dL (normal <0,500). Alle hereditären Thrombophilien waren normal. Zwei Wochen nach intravitrealer Injektion eines Dexamethason-Implantats (Ozurdex) verbesserte sich die Sehschärfe von 20/200 auf 20/40. Die schnelle Besserung unter Dexamethason könnte die Entzündungshypothese stützen.
Ntora et al. (2021) berichteten über den weltweit ersten Fall einer Papillophlebitis bei einer 22-jährigen Frau mit einer Kombination aus FVL-Heterozygotie und MTHFR-C677T-Heterozygotie-Mutation2). Eine vollständige Remission wurde allein mit oralem Methylprednisolon erreicht. Das genetische Screening erwies sich als wichtiger Suchparameter bei der Diagnose der Papillophlebitis. Eine hämatologische Konsultation gilt als wesentlich zur Vermeidung zukünftiger Komplikationen.
Insausti-García A, Reche-Sainz JA, Ruiz-Arranz C, López Vázquez Á, Ferro-Osuna M.. Papillophlebitis in a COVID-19 patient: Inflammation and hypercoagulable state. Eur J Ophthalmol. 2022;32(1):NP168-NP172. doi:10.1177/1120672120947591. PMID:32735134; PMCID:PMC7399568.
Ntora E, Dalianis G, Terzidou C. Papillophlebitis Associated With Coexisting Heterozygous Mutations of Factor V Leiden and Methylenetetrahydrofolate Reductase Enzyme-(C677T). Cureus. 2021;13(5):e15081. doi:10.7759/cureus.15081. PMID:34159001; PMCID:PMC8212846.
Abdel Jalil S, Amer R. The Spectrum of Papillophlebitis. Ocul Immunol Inflamm. 2024;32(10):2515-2520. PMID: 38842197.