良性型的組織學特徵
非畸胎瘤型:形成神經上皮樣管腔結構。
畸胎瘤型:除神經上皮樣結構外,還含有軟骨、橫紋肌等異位組織。
共同特徵:核異型性輕微,浸潤所見及核分裂像少見。

髓上皮瘤是一種罕見的眼內腫瘤,起源於睫狀體無色素上皮。睫狀體無色素上皮來源於胚胎神經管上皮,髓上皮瘤表現出反映其多向分化潛能的獨特組織學特徵。它曾一度被描述為與視網膜母細胞瘤相關的腫瘤。
最常見的發生部位是睫狀體,起源於視神經或視網膜的病例罕見。它主要發生於兒童,尤其是1至10歲,成人發病極為罕見。Kaliki等人對41例病例的分析顯示,診斷時的中位年齡為5歲1。這是一種非常罕見的腫瘤,難以確定確切發生率,在眼科臨床實踐中極為少見2。
臨床上最重要的挑戰是與同樣發生於同齡兒童且表現為白瞳症的視網膜母細胞瘤進行鑑別。確診通常依靠眼球摘除後的病理組織學檢查。
髓上皮瘤在早期通常無症狀,可能由家長偶然發現或在嬰幼兒健康檢查中因紅光反射異常而被發現。主要的發現契機如下所示。
在Kaliki等人對41例病例的分析中,續發性青光眼佔44%,虹膜新生血管佔51%,白內障佔46%,水晶體半脫位佔27%,白瞳症、續發性青光眼和水晶體異常被報告為特徵性的臨床三聯徵13。
散瞳後的前眼部檢查及隅角鏡檢查可發現睫狀體區域的白色至黃白色腫塊。腫塊表面不規則,可能含有囊狀結構。
良性型的組織學特徵
非畸胎瘤型:形成神經上皮樣管腔結構。
畸胎瘤型:除神經上皮樣結構外,還含有軟骨、橫紋肌等異位組織。
共同特徵:核異型性輕微,浸潤所見及核分裂像少見。
惡性型的組織學特徵
浸潤所見:明顯浸潤周圍組織。
核分裂像:出現大量核分裂像。
眼外浸潤風險:可能發生鞏膜外、眼眶浸潤及轉移。
腫瘤增大並向眼球外浸潤時,出現眼球突出及眼球運動障礙。也可能因續發性青光眼導致眼壓升高和角膜水腫。
髓上皮瘤的發生被認為源於睫狀體無色素上皮的胚胎期發育異常。正常發育中神經管上皮的形成過程被認為與腫瘤形成有關,但具體發病機制尚不明確。
尚未確定特定的環境風險因素。關於性別、地區及人種差異也缺乏明確數據。
在遺傳背景方面,部分病例報告了與DICER1基因突變的相關性。DICER1突變是導致胸膜肺母細胞瘤、腎臟腫瘤、甲狀腺腫瘤等多器官腫瘤的癌症易感症候群(DICER1症候群)的致病基因,已有報導指出睫狀體髓上皮瘤可作為伴有胸膜肺母細胞瘤的家族性腫瘤易感症候群的一種表現出現45。此外,在散發病例中,腫瘤組織中也發現了體細胞性DICER1突變,提示DICER1路徑異常可能參與腫瘤發生6。然而,大多數髓上皮瘤是散發性的,家族性發病僅限於少數病例。
在畸胎瘤樣型中,由於包含軟骨、橫紋肌等多種異位組織,因此認為多能細胞參與了腫瘤形成。這與睫狀體無色素上皮在胚胎期保持多向分化潛能是一致的。
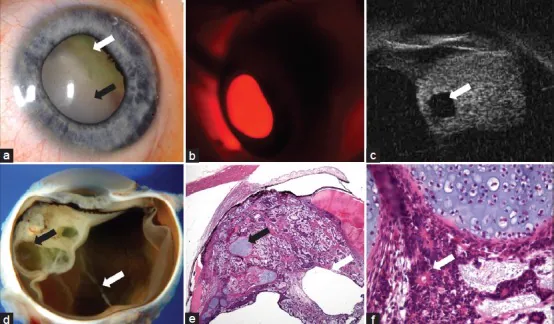
**超音波生物顯微鏡(UBM)**是確認和評估睫狀體腫塊形態最有用的檢查。它可以顯示與睫狀體相鄰的實性或囊實性腫塊,並評估腫塊範圍、與睫狀體的關係以及前段浸潤的存在。Kaliki等人的分析顯示,61%的病例中確認了腫瘤內囊腫,UBM顯示囊性結構是強烈提示本病的表現1。
MRI用於評估腫瘤範圍和眼外浸潤。當懷疑眼眶或視神經浸潤時,它為治療決策提供必要資訊。
CT掃描有助於評估鈣化的存在,並有助於與視網膜母細胞瘤鑑別(視網膜母細胞瘤常出現鈣化)。
眼底檢查和前段檢查在散瞳下使用裂隙燈顯微鏡和間接檢眼鏡進行。直接觀察睫狀體腫塊並評估視網膜合併病變(如漿液性視網膜剝離)。
確診通常通過眼球摘除後的病理組織學檢查進行。由於睫狀體活檢技術上困難且存在腫瘤播散風險,當臨床和影像學檢查強烈懷疑髓上皮瘤時,通常先行眼球摘除術。
組織病理學上,診斷為形成類似胚胎神經上皮的管狀、乳頭狀或片狀結構的上皮性腫瘤。畸胎樣型的特點是存在異位組織,如軟骨、橫紋肌和腦組織樣成分。
| 疾病 | 鑑別要點 |
|---|---|
| 視網膜母細胞瘤 | 特徵為白瞳、CT上鈣化、多發性。起源於視網膜。髓上皮瘤主要表現為睫狀體腫塊。 |
| 睫狀體黑色素細胞瘤 | 良性,色素豐富。多見於中老年人,UBM上呈高回聲腫塊。 |
| 睫狀體惡性黑色素瘤 | 發生於中老年人。色素性腫塊,有時伴有哨兵血管。 |
| 永存原始玻璃體增生症(PHPV) | 先天性。玻璃體內形成纖維血管膜,表現為白瞳。 |
眼球摘除後使用義眼。考慮到美觀和社交適應,通常早期植入眼眶植入物。
對於強烈提示為良性的小型腫瘤,可選擇通過睫狀體切除術進行局部切除。然而,手術進入睫狀體在技術上具有挑戰性,能夠實施的機構和醫生有限。此外,切除後仍有腫瘤復發風險,因此需要謹慎選擇病例並嚴格追蹤。
如果眼外浸潤進展,腫瘤累及眼眶,則需要行眼眶內容物剜除術。由於創傷大且術後美觀問題,需謹慎判斷適應症。
| 病理類型 | 預後特徵 |
|---|---|
| 良性型 | 眼球摘除後若無轉移,預後良好。局部復發率低。 |
| 惡性型 | 有眼外浸潤和遠端轉移的風險。一旦轉移,預後不良。 |
| 眼外浸潤病例 | 即使進行眼眶內容物剜除術後,也需注意局部復發和轉移。 |
遠端轉移相對少見,但惡性型和眼外浸潤病例需要充分追蹤。若發生轉移,可考慮化療、放療等治療,但尚未確立標準方案。
髓上皮瘤起源於睫狀體無色素上皮。睫狀體無色素上皮在胚胎期由神經管來源的神經上皮形成。因此,髓上皮瘤的組織學表現為類似胚胎期神經管上皮的管腔、乳頭和片狀結構。
非畸胎瘤型僅形成神經上皮樣的管腔和乳頭結構。是最接近正常神經管上皮形態的類型。細胞呈柱狀至立方狀,有時呈假複層排列。
畸胎瘤型除神經上皮樣結構外,還包含軟骨、橫紋肌、腦組織樣成分等多種組織。這反映了腫瘤起始細胞的高度多分化潛能,組織學類似畸胎瘤。良性畸胎瘤型核異型性和核分裂象少,增殖邊界清晰。
惡性型除神經上皮樣結構外,還可見以下表現:
這些發現是判斷惡性程度和預後預測的依據。也有報告指出存在良惡性界限模糊的中間型,病理診斷需要經驗。
在惡性類型中,腫瘤細胞通過鞏膜導水管(emissary canal)浸潤眼眶是主要途徑。血行轉移被認為是經由睫狀體豐富的血管網進行全身播散,但詳細機制仍在研究中。
DICER1基因編碼參與小RNA生物合成的RNase III酶。DICER1的胚系突變導致DICER1症候群,與胸膜肺母細胞瘤、腎腫瘤、甲狀腺腫瘤等多種腫瘤相關。近年來,有報告指出部分髓上皮瘤存在DICER1突變,眼腫瘤與DICER1症候群的關聯性備受關注。
識別DICER1突變對於選擇遺傳諮詢對象以及考慮家族遺傳篩檢的適應症可能很重要。最近對睫狀體髓上皮瘤的臨床病理學分析再次指出,結合分子遺傳學檢測的系統性評估的重要性7。
髓上皮瘤病例數少,難以進行大規模臨床研究。以Shields等人為代表的眼腫瘤專科機構積累的大規模病例系列,有助於完善良惡性的病理鑑別標準。核分裂像計數和浸潤發現的定量評估標準的標準化是未來的課題。
對於小型、良性型髓上皮瘤,局部切除(睫狀體切除術)的適應症擴大正在研究中。雖然有可能避免眼球摘除並保留視功能,但需要建立術後復發風險管理和長期追蹤方案。
畸胎樣型與非畸胎樣型在惡性化頻率、眼外浸潤風險和預後方面是否存在差異尚未完全闡明。期待通過大規模多中心研究進行比較分析。
Kaliki S, Shields CL, Eagle RC Jr, Vemuganti GK, Almeida A, Manjandavida FP, Mulay K, Honavar SG, Shields JA. Ciliary body medulloepithelioma: analysis of 41 cases. Ophthalmology. 2013;120(12):2552-2559. doi:10.1016/j.ophtha.2013.05.015. PMID: 23796765. ↩ ↩2 ↩3 ↩4
Tadepalli SH, Shields CL, Shields JA, Honavar SG. Intraocular medulloepithelioma - A review of clinical features, DICER 1 mutation, and management. Indian J Ophthalmol. 2019;67(6):755-762. doi:10.4103/ijo.IJO_845_19. PMID: 31124483; PMCID: PMC6552580. ↩ ↩2
Peshtani A, Kaliki S, Eagle RC, Shields CL. Medulloepithelioma: A triad of clinical features. Oman J Ophthalmol. 2014;7(2):93-95. doi:10.4103/0974-620X.137171. PMID: 25136238; PMCID: PMC4134557. ↩
Kramer GD, Arepalli S, Shields CL, Shields JA. Ciliary body medulloepithelioma association with pleuropulmonary blastoma in a familial tumor predisposition syndrome. J Pediatr Ophthalmol Strabismus. 2014;51:e48-e50. doi:10.3928/01913913-20140709-03. PMID: 25032694. ↩
Priest JR, Williams GM, Manera R, Jenkinson H, Bründler MA, Davis S, Murray TG, Galliani CA, Dehner LP. Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma—a report from the International Pleuropulmonary Blastoma Registry. Br J Ophthalmol. 2011;95(7):1001-1005. doi:10.1136/bjo.2010.189779. PMID: 21156700. ↩
Durieux E, Descotes F, Nguyen AM, Grange JD, Devouassoux-Shisheboran M. Somatic DICER1 gene mutation in sporadic intraocular medulloepithelioma without pleuropulmonary blastoma syndrome. Hum Pathol. 2015;46(5):783-787. doi:10.1016/j.humpath.2015.01.020. PMID: 25791583. ↩
August AH, Cernichiaro-Espinosa LA, Moctezuma-Davila M, Wibbelsman TD, Wilson MW, Chévez-Barrios P, Shields CL, Lally SE, Eberhart CG, Orr BA, Simpson E, Eagle RC, Milman T. Ciliary Body Medulloepithelioma: Clinical and Pathologic Challenges with a Focus on Molecular Genetics. Semin Ophthalmol. 2025;40(6):518-528. doi:10.1080/08820538.2025.2457066. PMID: 39869033. ↩