Osteoma
Frequência: 60-80% dos pacientes com FAP
Locais preferenciais: mandíbula, seios paranasais, crânio
Significado diagnóstico: ≥3 sugere fortemente GS. Pode preceder o diagnóstico de FAP em 17 anos

A síndrome de Gardner (GS) é uma variante fenotípica da polipose adenomatosa familiar (PAF). Além da polipose adenomatosa do cólon, caracteriza-se por tumores extracolônicos, como osteomas, tumores de tecidos moles (tumor desmoide) e cistos epidérmicos. O padrão de herança é autossômico dominante.
O gene causador é o gene APC (adenomatous polyposis coli), localizado no cromossomo 5q21. Mutações na linhagem germinativa causam a doença, com probabilidade de 50% de transmissão aos filhos. A prevalência é estimada em 1 a cada 100.000-160.000 pessoas 4).
A FAP tem os seguintes subtipos 1).
A síndrome de Gardner representa cerca de 1% de todos os cânceres colorretais 1). Os pólipos colorretais começam a aparecer por volta dos 10 anos de idade, chegando a centenas ou dezenas de milhares. Sem tratamento, 50% dos pacientes desenvolvem câncer colorretal aos 40 anos, e quase todos aos 60 anos. Mutações de novo ocorrem em cerca de 20-30% dos casos 1)5).
Mutações de novo (mutações novas) no gene APC ocorrem em cerca de 20-30% dos casos, portanto a doença pode surgir mesmo sem histórico familiar1)5). É importante notar que a ausência de histórico familiar não exclui a SG.
Os pólipos colorretais geralmente são assintomáticos, sendo frequentemente descobertos tardiamente. Quando os sintomas aparecem, os principais são os seguintes.
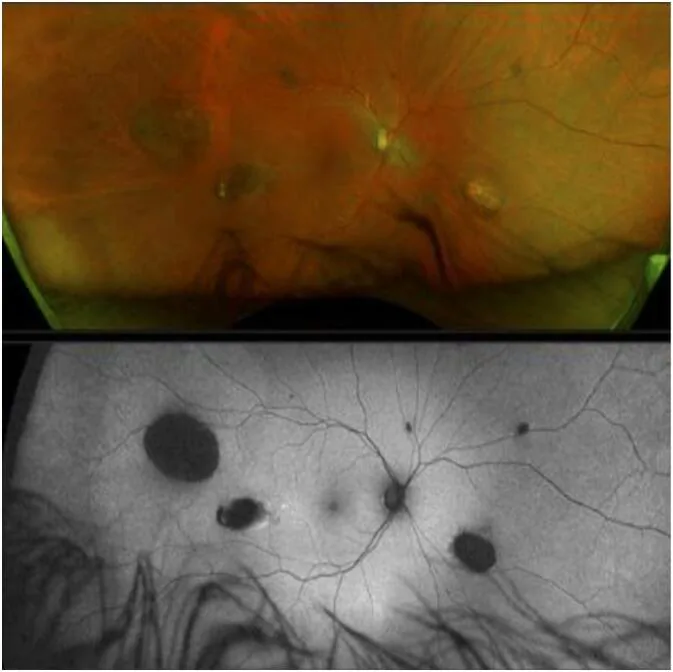
CHRPE (hipertrofia congênita do epitélio pigmentar da retina) é o sinal extra-colônico mais precoce da GS, presente desde o nascimento 1).
No entanto, a GS não pode ser descartada mesmo na ausência de CHRPE 2).
Osteoma
Frequência: 60-80% dos pacientes com FAP
Locais preferenciais: mandíbula, seios paranasais, crânio
Significado diagnóstico: ≥3 sugere fortemente GS. Pode preceder o diagnóstico de FAP em 17 anos
CHRPE
Frequência: 74% dos pacientes com FAP
Características: bilateral, múltiplo, em forma de ervilha. Presente desde o nascimento, o sinal mais precoce
Nota: GS não pode ser descartado mesmo sem CHRPE
Cisto Epidérmico
Frequência: cerca de 70%
Locais preferenciais: cabeça e pescoço
Características: aparece desde jovem, frequentemente múltiplo
Tumor Desmoide
Frequência: 12-15% dos pacientes com FAP
Locais preferenciais: Abdome (37-50%), seguido por cintura escapular e parede torácica
Atenção: Segunda causa de morte após câncer colorretal em pacientes com FAP. Mortalidade de 10-50%
Presentes em 30-75% dos pacientes com FAP1). Dentes impactados e supranumerários (11-27% em pacientes com FAP vs 0-4% na população geral)1).
O risco de vários tumores malignos fora do cólon também aumenta.
| Carcinoma | Risco ao longo da vida |
|---|---|
| Câncer de tireoide | PAF 2–12%, tipo Gardner 10% |
| Câncer de intestino delgado | 4–12% |
| Câncer de pâncreas | Cerca de 1% |
(Fonte: Referência 1)1)
CHRPE esporádico é encontrado na população geral com frequência de 1,2 a 4,4%, portanto a presença isolada de CHRPE não permite diagnosticar síndrome de Gardner. CHRPE associado à síndrome de Gardner caracteriza-se por ser bilateral, múltiplo, em forma de ervilha e com bordas irregulares. Por outro lado, a síndrome de Gardner não pode ser descartada mesmo na ausência de CHRPE2).
A causa da GS é uma mutação germinativa no gene APC (5q21). A maioria das mutações são frameshift ou nonsense, resultando em proteína APC truncada (perda de função).
Há uma correlação clara entre o local da mutação e o fenótipo 1).
| Local da mutação | Fenótipo associado |
|---|---|
| Códon 311–1444 | CHRPE |
| Códon 767–1578 | Osteoma |
| Éxon 1399 em diante | Desmoide (risco 800 vezes maior) |
| Códon 1309 | Câncer colorretal precoce aos 20 anos |
A região de cluster de mutação (mutation cluster region; MCR) são os códons 1250–1464 1), e a mutação mais frequente é no códon 1309 (cerca de 10% do total), seguida pelo códon 1061 (cerca de 5%) 1). A idade de início do câncer colorretal também varia conforme o local da mutação: no códon 1309, cerca de 20 anos; nos códons 168–1580, cerca de 30 anos; e nos demais, cerca de 52 anos 1).
O teste genético molecular do gene APC pode ser realizado desde a infância. A colonoscopia é frequentemente recomendada a partir dos 10 anos de idade. Se uma mutação APC for conhecida na família, é importante confirmar a presença ou ausência da mutação por teste genético.
O diagnóstico definitivo é feito por teste genético molecular do gene APC. Recomenda-se o teste de painel de sequenciamento de nova geração (NGS) 1).
O teste genético molecular do gene APC é a base para o diagnóstico definitivo da síndrome de Gardner, mas em alguns pacientes a mutação pode não ser detectada. O uso de painéis NGS melhorou a taxa de detecção 1). Os resultados do teste genético são interpretados em conjunto com os achados clínicos.
A CHRPE em si não se maligniza e não afeta a função visual. Não é necessária intervenção oftalmológica, apenas observação periódica.
Usado antes da cirurgia ou em casos leves para inibir o crescimento de pólipos.
A farmacoterapia isolada não pode eliminar o risco de câncer colorretal.
A colectomia é recomendada antes dos 25 anos, idealmente entre os 16 e 20 anos2). Existem três tipos principais de procedimentos2).
Colectomia Total com Ileostomia Permanente
Característica: Procedimento mais definitivo. Nenhum epitélio colorretal remanescente
Vantagem: Redução máxima do risco de câncer colorretal
Desvantagens: Requer estoma permanente, afetando a qualidade de vida
Proctocolectomia total restauradora com anastomose bolsa ileal-anal
Características: Técnica que preserva a continuidade intestinal
Vantagens: Evita estoma permanente
Desvantagens: Risco de complicações urológicas e sexuais
Colectomia total com anastomose ileorretal
Características: Menos invasivo, adequado para pacientes jovens
Vantagens: Invasão cirúrgica relativamente pequena
Desvantagens: A vigilância do reto remanescente é necessária continuamente
O tumor desmoide é histologicamente benigno, mas altamente infiltrativo localmente e propenso a recorrência. O tratamento requer uma abordagem multidisciplinar 4)5).
Após a colectomia, a seguinte vigilância é continuada 2).
Depende do número de pólipos e do grau de displasia. Se houver poucos pólipos, pode-se acompanhar com medicamentos como AINEs ou inibidores de COX-2. No entanto, se houver 20 ou mais pólipos ou displasia de alto grau, a ressecção profilática é recomendada. A idade ideal para cirurgia é entre 16 e 20 anos 2).
A proteína APC é um supressor tumoral composto por 2800 aminoácidos. Sua estrutura de domínios principais inclui uma região de oligomerização, repetições armadillo, região de ligação à β-catenina, região de ligação à axina e região de ligação aos microtúbulos 1).
Mutações no APC causam ativação constitutiva da via de sinalização Wnt. A proteína APC normal fosforila a β-catenina e promove sua degradação no proteassoma. Na mutação do APC, essa função é perdida, a β-catenina se acumula no citoplasma e forma um complexo com o fator de transcrição TCF/LEF no núcleo, promovendo a transcrição de genes de proliferação celular.
A maioria das mutações são mutações de deslocamento de quadro ou nonsense, resultando em uma proteína APC truncada (perda de função) 1).
Em tumores desmoides esporádicos, mutações no CTNNB1 (gene da β-catenina) são encontradas em mais de 75% dos casos, e a mesma via está envolvida 3).
A CHRPE ocorre como um distúrbio de proliferação celular localizada no epitélio pigmentar da retina. Nas áreas onde o epitélio pigmentar da retina prolifera anormalmente, o pigmento se concentra e hipertrofia, dando uma aparência característica em contraste com as áreas lacunares (despigmentadas) circundantes. Praticamente não há potencial de malignização e nenhum efeito na função visual.
O exame de painel de sequenciamento de próxima geração (NGS) está melhorando a precisão na identificação de locais de mutação no gene APC1). A estratificação de risco com base na correlação genótipo-fenótipo está avançando, e espera-se a construção de estratégias de vigilância e tratamento personalizadas de acordo com o local da mutação1).
Litchinko et al. (2022) relataram um caso raro de grande tumor desmoide (170 mm) originado no pâncreas3). O manejo multidisciplinar foi realizado, demonstrando a dificuldade em determinar indicação e momento cirúrgico no desmoide pancreático. A eficácia do sorafenibe como terapia medicamentosa está sendo observada, e sua aplicação em casos de difícil ressecção cirúrgica está sendo considerada.
A aplicação do ultrassom focalizado de alta intensidade (HIFU) como terapia não invasiva para desmoides também está em fase de pesquisa3).
Albuquerque et al. (2025) relataram um caso de tumor desmoide pediátrico que exigiu reconstrução da função da articulação do ombro5). Uma abordagem multidisciplinar incluindo quimioterapia pré-operatória com vimblastina e metotrexato alcançou preservação funcional e controle tumoral. Desmoides do cíngulo do membro superior e parede torácica representam 37-50% dos casos, destacando a importância da reconstrução funcional em casos pediátricos.
Diaz et al. (2025) relataram um caso de síndrome de Gardner (GS) apresentando dilema cirúrgico incluindo carcinoma basocelular infiltrativo e complicações de artroplastia total do joelho, demonstrando a necessidade de manejo multidisciplinar para complicações raras4). Com prevalência de 1 em 100.000-160.000 pessoas, a importância da medicina personalizada está aumentando nesta doença.