Ostéome
Fréquence : 60 à 80 % des patients atteints de PAF
Sites de prédilection : Maxillaire, sinus paranasaux, crâne
Signification diagnostique : 3 ou plus suggèrent fortement un GS. Peut précéder le diagnostic de FAP de 17 ans

Le syndrome de Gardner (GS) est une variante phénotypique de la polypose adénomateuse familiale (PAF). En plus de la polypose adénomateuse du côlon, il se caractérise par des manifestations extracoliques telles que des ostéomes, des tumeurs des tissus mous (tumeurs desmoïdes) et des kystes épidermiques. La transmission est autosomique dominante.
Le gène responsable est le gène APC (adenomatous polyposis coli), situé sur le chromosome 5 en 5q21. La maladie est causée par une mutation germinale, avec un risque de transmission à la descendance de 50 %. La prévalence est estimée à 1 pour 100 000 à 160 000 personnes4).
La FAP comprend les sous-types suivants 1).
La PAF représente environ 1 % de tous les cancers colorectaux1). Les polypes coliques apparaissent vers l’âge de 10 ans et peuvent atteindre plusieurs centaines à plusieurs dizaines de milliers. Sans traitement, 50 % des patients développent un cancer colorectal à 40 ans, et presque tous à 60 ans. Des mutations de novo sont observées dans environ 20 à 30 % des cas1)5).
Des mutations de novo du gène APC surviennent dans environ 20 à 30 % des cas, ce qui signifie que la maladie peut survenir même en l’absence d’antécédents familiaux1)5). Il est important de noter que l’absence d’antécédents familiaux n’exclut pas le syndrome de Gardner.
Les polypes colorectaux sont généralement asymptomatiques, ce qui retarde souvent leur découverte. Lorsque des symptômes apparaissent, ils sont principalement les suivants.
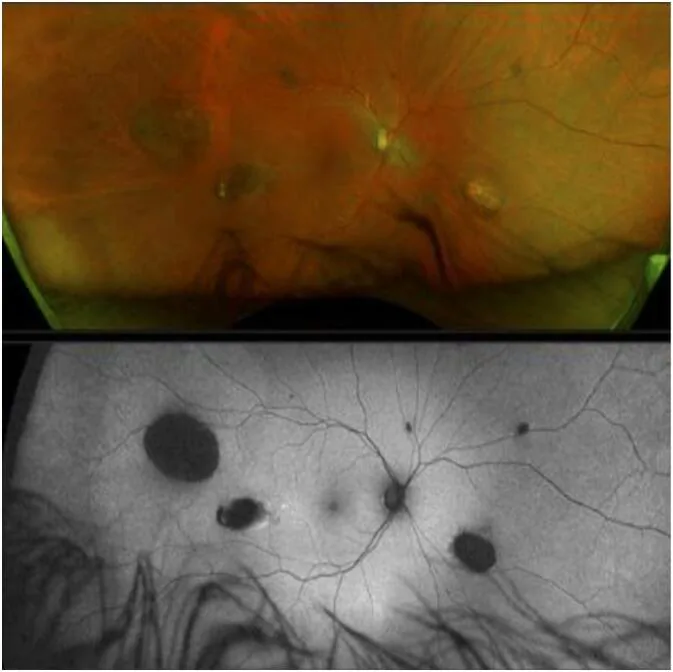
La CHRPE (hypertrophie congénitale de l’épithélium pigmentaire rétinien) est la manifestation extracolique la plus précoce du syndrome de Gardner, présente dès la naissance 1).
Cependant, l’absence de CHRPE n’exclut pas le GS2).
Ostéome
Fréquence : 60 à 80 % des patients atteints de PAF
Sites de prédilection : Maxillaire, sinus paranasaux, crâne
Signification diagnostique : 3 ou plus suggèrent fortement un GS. Peut précéder le diagnostic de FAP de 17 ans
CHRPE
Fréquence : 74 % des patients atteints de FAP
Caractéristiques : bilatéral, multiple, en forme de pois. Signe le plus précoce présent dès la naissance
Attention : l’absence de CHRPE n’exclut pas la GS
Kyste épidermique
Fréquence : environ 70%
Localisation préférentielle : tête et cou
Caractéristiques : apparaît dès le jeune âge, souvent multiple
Tumeur desmoïde
Fréquence : 12 à 15 % des patients atteints de PAF
Sites de prédilection : abdomen (37 à 50 %), puis ceinture scapulaire et paroi thoracique
Attention : Deuxième cause de décès chez les patients atteints de PAF après le cancer colorectal. Taux de mortalité de 10 à 50 %.
Observées chez 30 à 75 % des patients atteints de PAF1). Dents incluses et surnuméraires (11 à 27 % des patients PAF contre 0 à 4 % dans la population générale)1).
Le risque de divers cancers autres que le cancer colorectal est également augmenté.
| Cancer | Risque au cours de la vie |
|---|---|
| Cancer de la thyroïde | FAP 2–12 %, type Gardner 10 % |
| Cancer de l’intestin grêle | 4 à 12 % |
| Cancer du pancréas | Environ 1 % |
(Source : référence 1)1)
La CHRPE sporadique est observée dans 1,2 à 4,4 % de la population générale, donc la seule présence de CHRPE ne permet pas de diagnostiquer un syndrome de Gardner. La CHRPE associée au GS se caractérise par une atteinte bilatérale, multiple, en forme de pois et aux contours irréguliers. Inversement, l’absence de CHRPE n’exclut pas le GS2).
La cause du GS est une mutation germinale du gène APC (5q21). La majorité des mutations sont des décalages du cadre de lecture ou des mutations non-sens, produisant une protéine APC tronquée (perte de fonction).
Il existe une corrélation claire entre le site de mutation et le phénotype 1).
| Site de mutation | Phénotype associé |
|---|---|
| Codons 311-1444 | CHRPE |
| Codons 767-1578 | Ostéome |
| Exon 1399 et au-delà | Desmoïde (risque multiplié par 800) |
| Codon 1309 | Cancer colorectal précoce à 20 ans |
La région de cluster de mutation (mutation cluster region; MCR) se situe entre les codons 1250 et 14641), la mutation la plus fréquente étant au codon 1309 (environ 10% du total), suivie du codon 1061 (environ 5%)1). L’âge d’apparition du cancer colorectal varie également selon le site de mutation : environ 20 ans pour le codon 1309, autour de 30 ans pour les codons 168 à 1580, et environ 52 ans pour les autres1).
Le test de génétique moléculaire du gène APC peut être réalisé dès l’enfance. La coloscopie est souvent recommandée à partir de l’âge de 10 ans environ. Si une mutation APC est connue dans la famille, il est important de confirmer la présence ou l’absence de la mutation par un test génétique.
Le diagnostic définitif repose sur le test de génétique moléculaire du gène APC. Un test par panel de séquençage de nouvelle génération (NGS) est recommandé 1).
Le test de génétique moléculaire du gène APC permet de confirmer le diagnostic de GS, mais chez certains patients, la mutation peut ne pas être détectée. L’utilisation de panels de séquençage de nouvelle génération (NGS) a amélioré le taux de détection 1). Les résultats des tests génétiques doivent être interprétés en combinaison avec les observations cliniques.
La CHRPE elle-même ne devient pas maligne et n’affecte pas la fonction visuelle. Aucune intervention ophtalmique n’est nécessaire, seule une surveillance régulière est effectuée.
Utilisé avant la chirurgie ou dans les cas bénins pour inhiber la croissance des polypes.
Le traitement médicamenteux seul ne peut pas éliminer le risque de cancer colorectal.
La colectomie est recommandée avant l’âge de 25 ans, idéalement entre 16 et 20 ans2). Les trois principales techniques chirurgicales sont les suivantes2).
Colectomie totale avec iléostomie définitive
Caractéristiques : Technique la plus radicale. Aucun épithélium colique résiduel.
Avantages : Réduction maximale du risque de cancer colorectal.
Inconvénient : nécessité d’une stomie permanente, ce qui affecte la qualité de vie
Colectomie totale avec anastomose iléo-anale et réservoir iléal
Caractéristique : technique préservant la continuité intestinale
Avantage : permet d’éviter une stomie permanente
Inconvénient : risque de complications urologiques et sexuelles
Colectomie totale + anastomose iléo-rectale
Caractéristique : peu invasive, adaptée aux jeunes patients
Avantage : invasivité chirurgicale relativement faible
Inconvénient : une surveillance continue du rectum restant est nécessaire
Les tumeurs desmoïdes sont histologiquement bénignes, mais elles sont très infiltrantes localement et récidivent fréquemment. Le traitement nécessite une approche multidisciplinaire impliquant plusieurs spécialités4)5).
Après une résection colique, la surveillance suivante doit être poursuivie2).
Cela dépend du nombre de polypes et du degré de dysplasie. Pour un petit nombre de polypes, un traitement médicamenteux par AINS ou inhibiteurs de la COX-2 peut être envisagé. Cependant, si plus de 20 polypes ou des polypes avec dysplasie sévère sont présents, une résection prophylactique est recommandée. L’âge idéal pour la chirurgie est considéré comme étant entre 16 et 20 ans 2).
La protéine APC est un suppresseur de tumeur composé de 2800 acides aminés. Ses principaux domaines structuraux comprennent une région d’oligomérisation, des répétitions armadillo, une région de liaison à la β-caténine, une région de liaison à l’axine et une région de liaison aux microtubules 1).
Les mutations de l’APC entraînent une activation constitutive de la voie de signalisation Wnt. La protéine APC normale phosphoryle la β-caténine, favorisant sa dégradation par le protéasome. En cas de mutation de l’APC, cette fonction est perdue, la β-caténine s’accumule dans le cytoplasme, forme un complexe avec les facteurs de transcription TCF/LEF dans le noyau et active la transcription de gènes de prolifération cellulaire.
La majorité des mutations sont des décalages du cadre de lecture ou des mutations non-sens, produisant une protéine APC tronquée (perte de fonction) 1).
Dans les tumeurs desmoïdes sporadiques, des mutations de CTNNB1 (gène de la β-caténine) sont observées dans plus de 75 % des cas, impliquant la même voie 3).
La CHRPE résulte d’une prolifération cellulaire localisée anormale de l’épithélium pigmentaire rétinien. Dans les zones de prolifération anormale, le pigment se concentre et s’hypertrophie, créant un aspect caractéristique par contraste avec les lacunes environnantes (zones de dépigmentation). Le potentiel de malignité est quasiment nul et il n’y a pas d’impact sur la fonction visuelle.
Les tests de séquençage de nouvelle génération (NGS) améliorent la précision de l’identification des sites de mutation du gène APC1). La stratification des risques basée sur la corrélation génotype-phénotype progresse, et on espère développer des stratégies de surveillance et de traitement personnalisées en fonction du site de mutation1).
Litchinko et al. (2022) ont rapporté un rare cas de tumeur desmoïde volumineuse (170 mm) survenue dans le pancréas3). Une prise en charge multidisciplinaire par une équipe pluridisciplinaire a été réalisée, soulignant la difficulté de l’indication et du moment de la chirurgie dans les desmoïdes pancréatiques. L’efficacité du sorafénib en tant que traitement médicamenteux attire l’attention, et son application est envisagée pour les cas où la résection chirurgicale est difficile.
L’application des ultrasons focalisés de haute intensité (HIFU) pour le traitement non invasif des desmoïdes est également en phase de recherche3).
Albuquerque et al. (2025) ont rapporté un cas de tumeur desmoïde pédiatrique ayant nécessité une reconstruction fonctionnelle de l’articulation de l’épaule5). Une approche multidisciplinaire incluant une chimiothérapie préopératoire par vinblastine et méthotrexate a permis de préserver la fonction et de contrôler la tumeur. Les desmoïdes de la ceinture scapulaire et de la paroi thoracique représenteraient 37 à 50 % de l’ensemble, soulignant l’importance de la reconstruction fonctionnelle chez l’enfant.
Diaz et al. (2025) ont rapporté des cas de GS présentant des dilemmes chirurgicaux, y compris un carcinome basocellulaire invasif et une prothèse totale du genou, soulignant la nécessité d’une prise en charge multidisciplinaire des complications rares4). Dans cette maladie, dont la prévalence est estimée à 1 pour 100 000 à 160 000 personnes, l’importance de la médecine personnalisée augmente.