Osteoma
Frequenza: 60-80% dei pazienti con FAP
Sedi preferenziali: mandibola, seni paranasali, cranio
Significato diagnostico: 3 o più suggeriscono fortemente GS. Possono precedere la diagnosi di FAP di 17 anni

La sindrome di Gardner (GS) è una variante fenotipica della poliposi adenomatosa familiare (FAP). Oltre alla poliposi adenomatosa del colon, è caratterizzata da manifestazioni extracoliche come osteomi, tumori dei tessuti molli (tumori desmoidi) e cisti epidermiche. La trasmissione è autosomica dominante.
Il gene responsabile è il gene APC (adenomatous polyposis coli), situato sul cromosoma 5q21. La malattia è causata da mutazioni germinali e la probabilità di trasmissione alla prole è del 50%. La prevalenza è stimata in 1 caso ogni 100.000-160.000 persone4).
La FAP presenta i seguenti sottotipi1).
La FAP rappresenta circa l’1% di tutti i tumori del colon-retto1). I polipi del colon compaiono intorno ai 10 anni e possono raggiungere da centinaia a decine di migliaia. Senza trattamento, il 50% dei pazienti sviluppa un cancro del colon-retto entro i 40 anni e quasi tutti entro i 60 anni. Le mutazioni de novo si verificano in circa il 20-30% dei casi1)5).
Poiché le mutazioni de novo del gene APC si verificano con una frequenza di circa il 20-30%, la sindrome può manifestarsi anche in assenza di storia familiare1)5). È importante notare che l’assenza di storia familiare non esclude la GS.
I polipi del colon-retto sono solitamente asintomatici e spesso vengono scoperti tardivamente. Quando compaiono sintomi, questi sono principalmente i seguenti.
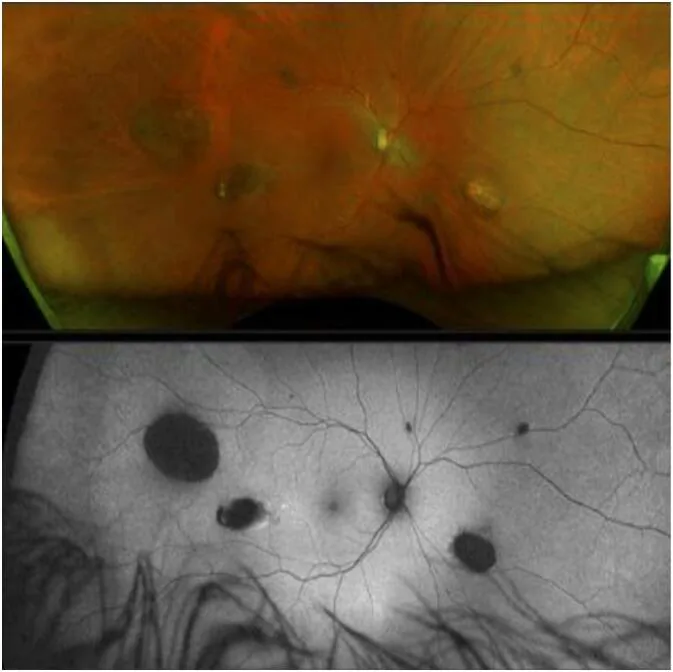
La CHRPE (ipertrofia congenita dell’epitelio pigmentato retinico) è la più precoce manifestazione extra-colica della GS ed è presente dalla nascita 1).
Tuttavia, la GS non può essere esclusa anche in assenza di CHRPE2).
Osteoma
Frequenza: 60-80% dei pazienti con FAP
Sedi preferenziali: mandibola, seni paranasali, cranio
Significato diagnostico: 3 o più suggeriscono fortemente GS. Possono precedere la diagnosi di FAP di 17 anni
CHRPE
Frequenza: 74% dei pazienti con FAP
Caratteristiche: bilaterali, multipli, a forma di pisello. Sono il segno più precoce, presente dalla nascita
Nota: l’assenza di CHRPE non esclude GS
Cisti epidermoide
Frequenza: circa 70%
Sedi preferenziali: testa e collo
Caratteristiche: compare in giovane età, spesso si osservano più lesioni
Tumore desmoide
Frequenza: 12-15% dei pazienti con FAP
Sedi preferenziali: addome (37-50%), seguito da cingolo scapolare e parete toracica
Attenzione: seconda causa di morte nei pazienti con FAP dopo il carcinoma del colon-retto. Mortalità 10-50%
Presenti nel 30-75% dei pazienti con FAP1). Si osservano denti inclusi e sovrannumerari (11-27% nei pazienti con FAP rispetto allo 0-4% nella popolazione generale)1).
Il rischio di vari tumori maligni aumenta anche al di fuori del colon.
| Tumore | Rischio nel corso della vita |
|---|---|
| Cancro della tiroide | FAP 2-12%, tipo Gardner 10% |
| Cancro dell’intestino tenue | 4-12% |
| Cancro del pancreas | Circa 1% |
(Fonte: citazione 1) 1)
La CHRPE sporadica si osserva anche nella popolazione generale con una frequenza dell’1,2-4,4%, quindi non si può diagnosticare la GS solo per la presenza di CHRPE. La CHRPE associata a GS è caratterizzata da bilateralità, multipla, a forma di pisello e con bordi irregolari. Al contrario, la GS non può essere esclusa anche in assenza di CHRPE 2).
La causa della GS è una mutazione germinale del gene APC (5q21). La maggior parte delle mutazioni sono frameshift o non senso, che portano alla produzione di una proteina APC tronca (con perdita di funzione).
Esiste una chiara correlazione tra il sito della mutazione e il fenotipo 1).
| Sito della mutazione | Fenotipo associato |
|---|---|
| Codoni 311-1444 | CHRPE |
| Codoni 767-1578 | Osteoma |
| Esone 1399 e successivi | Desmoide (rischio 800 volte maggiore) |
| Codone 1309 | Cancro del colon-retto precoce a 20 anni |
La regione del cluster di mutazione (mutation cluster region; MCR) comprende i codoni 1250-1464 1), la mutazione più frequente è al codone 1309 (circa il 10% del totale), seguita dal codone 1061 (circa il 5%) 1). Anche l’età di insorgenza del cancro del colon-retto varia in base al sito di mutazione: per il codone 1309 è di circa 20 anni, per i codoni 168-1580 è intorno ai 30 anni, mentre per le altre mutazioni è di circa 52 anni 1).
Il test genetico molecolare del gene APC può essere eseguito fin dall’infanzia. La colonscopia è spesso raccomandata a partire dall’età di circa 10 anni. Se una mutazione APC è nota in famiglia, è importante confermare la presenza o l’assenza della mutazione tramite test genetico.
La diagnosi definitiva si basa sul test genetico molecolare del gene APC. Si raccomanda il test con pannello di sequenziamento di nuova generazione (NGS) 1).
Il test genetico molecolare del gene APC fornisce la base per la diagnosi definitiva di GS, ma in alcuni pazienti la mutazione potrebbe non essere rilevata. L’uso di pannelli NGS ha migliorato il tasso di rilevamento 1). I risultati dei test genetici devono essere valutati in combinazione con i reperti clinici.
La CHRPE di per sé non è maligna e non influisce sulla funzione visiva. Non è necessario alcun intervento oftalmologico, solo un monitoraggio regolare.
Viene utilizzata prima dell’intervento chirurgico o nei casi lievi per inibire la crescita dei polipi.
La terapia farmacologica da sola non può eliminare il rischio di cancro del colon-retto.
La colectomia è raccomandata entro i 25 anni, idealmente tra i 16 e i 20 anni2). Le principali tecniche chirurgiche sono le seguenti tre tipologie2).
Colectomia totale con ileostomia permanente
Caratteristiche: la tecnica più radicale. Non rimane epitelio del colon
Vantaggi: riduce al massimo il rischio di cancro del colon
Svantaggio: richiede una stomia permanente, con impatto sulla qualità della vita
Colectomia totale proctocolectomia restaurativa con anastomosi ileo-anale
Caratteristica: tecnica che preserva la continuità intestinale
Vantaggio: evita una stomia permanente
Svantaggi: rischio di complicanze urologiche e sessuali
Colectomia totale + anastomosi ileo-rettale
Caratteristiche: meno invasiva, adatta a pazienti giovani
Vantaggi: invasività chirurgica relativamente ridotta
Svantaggio: è necessaria una sorveglianza continua del retto residuo
I tumori desmoidi sono istologicamente benigni, ma altamente infiltranti localmente e tendono a recidivare. Il trattamento richiede un approccio multidisciplinare 4)5).
大腸切除後も以下のサーベイランスを継続する2)。
ポリープの数や異形成の程度によって異なる。少数のポリープであればNSAIDsやCOX-2阻害薬による薬物療法で経過をみることがある。しかし、20個以上や高度異形成ポリープが認められる場合は予防的切除が推奨される。理想的な手術時期は16〜20歳とされている2)。
APCタンパク質は2800アミノ酸からなる癌抑制因子である。主要なドメイン構造として、オリゴマー化領域・アルマジロリピート・β-カテニン結合領域・アキシン結合領域・微小管結合領域を持つ1)。
APC変異はWntシグナル伝達経路の恒常的活性化を引き起こす。正常なAPCタンパク質はβ-カテニンをリン酸化してプロテアソームでの分解を促進する。APC変異ではこの機能が失われ、β-カテニンが細胞質に蓄積し、核内でTCF/LEF転写因子と複合体を形成して細胞増殖遺伝子の転写を促進する。
変異の大半はフレームシフトまたはナンセンス変異であり、切断型(機能喪失型)APCタンパク質が産生される1)。
散発性デスモイド腫瘍では、CTNNB1(β-カテニン遺伝子)変異が75%以上に認められ、同様の経路が関与する3)。
CHRPEは網膜色素上皮の局所的な細胞増殖異常として生じる。網膜色素上皮が異常増殖した部分は色素が濃縮・肥大し、周囲のラクナ(脱色素化領域)とのコントラストで特徴的な外観を呈する。悪性化ポテンシャルはほぼなく、視機能への影響も認められない。
次世代シークエンシング(NGS)パネル検査により、APC遺伝子の変異部位の同定精度が向上している1)。遺伝子型と表現型の相関に基づくリスク層別化が進み、変異部位に応じた個別化サーベイランス・治療戦略の構築が期待されている1)。
Litchinkoら(2022)は、膵臓に発生した大型デスモイド腫瘍(170 mm)の稀な1例を報告した3)。多学科チームによる集学的管理が行われ、膵臓デスモイドにおける手術適応・タイミングの難しさを提示した。薬物療法としてソラフェニブの有効性が注目されており、外科切除が困難な症例への適用が検討されている。
高密度焦点式超音波(HIFU)の非侵襲的デスモイド治療への応用も研究段階にある3)。
Albuquerqueら(2025)は、肩関節機能再建を要した小児デスモイド腫瘍の1例を報告した5)。ビンブラスチン+メトトレキサートによる術前化学療法を含む集学的アプローチにより、機能温存と腫瘍制御を達成した。肩甲帯・胸壁デスモイドが全体の37〜50%を占めるとされ、小児例における機能再建の重要性を示している。
Diazら(2025)は、浸潤性基底細胞癌と全膝関節置換術合併例を含む外科的ジレンマを呈するGS症例を報告し、稀な合併症への集学的対応が求められることを示した4)。有病率が10万〜16万人に1人とされる本疾患において、個別化医療の重要性が増している。