استئوم
فراوانی: 60-80% از بیماران FAP
محلهای شایع: استخوان فک، سینوسها، جمجمه
اهمیت تشخیصی: وجود سه یا بیشتر به شدت نشاندهنده GS است. ممکن است 17 سال قبل از تشخیص FAP ظاهر شود

سندرم گاردنر (Gardner syndrome; GS) یکی از انواع فنوتیپی پولیپوز آدنوماتوز خانوادگی (familial adenomatous polyposis; FAP) است. علاوه بر پولیپوز آدنوماتوز روده بزرگ، با تومورهای خارج رودهای مانند استئوما، تومورهای بافت نرم (تومور دسموئید) و کیستهای اپیدرمی مشخص میشود. الگوی وراثت اتوزومال غالب (که قبلاً اتوزومال غالب نامیده میشد) دارد.
ژن عامل، ژن APC (adenomatous polyposis coli) است که در موقعیت 5q21 کروموزوم 5 قرار دارد. جهش در سلولهای زایا باعث بروز بیماری میشود و احتمال انتقال به فرزند ۵۰٪ است. شیوع آن ۱ در ۱۰۰٬۰۰۰ تا ۱۶۰٬۰۰۰ نفر گزارش شده است4).
FAP زیرگروههای زیر را دارد 1).
FAP حدود 1% از کل سرطانهای روده بزرگ را تشکیل میدهد 1). پولیپهای روده بزرگ از حدود 10 سالگی ظاهر شده و به صدها تا دهها هزار عدد میرسند. در صورت عدم درمان، 50% در 40 سالگی و تقریباً همه موارد در 60 سالگی به سرطان روده بزرگ مبتلا میشوند. جهش de novo (جهش جدید) در حدود 20-30% موارد مشاهده میشود 1)5).
جهشهای de novo (جهشهای جدید) در ژن APC با فراوانی حدود ۲۰ تا ۳۰٪ رخ میدهد، بنابراین حتی بدون سابقه خانوادگی نیز ممکن است بیماری بروز کند1)5). توجه به این نکته ضروری است که عدم وجود سابقه خانوادگی به تنهایی نمیتواند GS را رد کند.
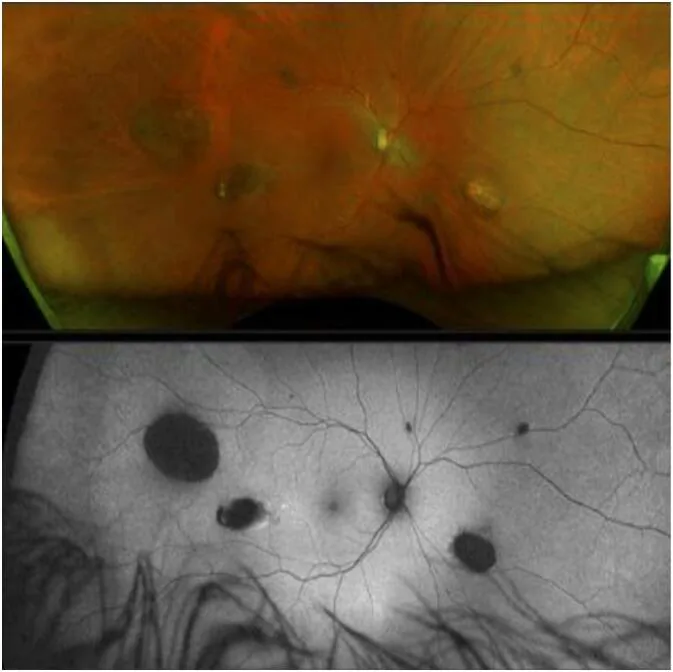
پولیپهای روده بزرگ معمولاً بدون علامت هستند و اغلب دیر تشخیص داده میشوند. در صورت بروز علائم، موارد زیر غالب هستند.
CHRPE (هیپرتروفی مادرزادی اپیتلیوم رنگدانهدار شبکیه) اولین علامت خارج روده ای GS است که از بدو تولد وجود دارد 1).
با این حال، عدم وجود CHRPE GS را رد نمیکند 2).
استئوم
فراوانی: 60-80% از بیماران FAP
محلهای شایع: استخوان فک، سینوسها، جمجمه
اهمیت تشخیصی: وجود سه یا بیشتر به شدت نشاندهنده GS است. ممکن است 17 سال قبل از تشخیص FAP ظاهر شود
CHRPE
فراوانی: 74% از بیماران FAP
ویژگی: دوطرفه، متعدد، به شکل نخود. اولین علامت که از بدو تولد وجود دارد
توجه: عدم وجود CHRPE نیز نمیتواند GS را رد کند
کیست اپیدرمی
فراوانی: حدود ۷۰٪
محل شایع: سر و گردن
ویژگی: از سنین پایین ظاهر میشود و اغلب چندتایی دیده میشود
تومور دسموئید
فراوانی: ۱۲ تا ۱۵ درصد از بیماران FAP
محل شایع: شکم (۳۷ تا ۵۰ درصد)، سپس کمربند شانهای و دیواره قفسه سینه
توجه: دومین علت مرگ در بیماران FAP پس از سرطان روده بزرگ. میزان مرگومیر ۱۰ تا ۵۰ درصد
در ۳۰ تا ۷۵ درصد از بیماران FAP مشاهده میشود1). دندانهای نهفته و اضافی (در ۱۱ تا ۲۷ درصد از بیماران FAP در مقابل ۰ تا ۴ درصد در جمعیت عمومی) دیده میشود1).
خطر ابتلا به انواع تومورهای بدخیم علاوه بر روده بزرگ نیز افزایش مییابد.
| نوع سرطان | خطر در طول عمر |
|---|---|
| سرطان تیروئید | FAP ۲-۱۲٪، نوع گاردنر ۱۰٪ |
| سرطان روده کوچک | ۴-۱۲٪ |
| سرطان پانکراس | حدود ۱٪ |
(منبع: نقلقول ۱)1)
علت GS جهش در سلولهای زایای ژن APC (5q21) است. بیشتر جهشها از نوع تغییر چارچوب یا بیمعنی هستند که منجر به تولید پروتئین APC کوتاهشده (غیرفعال) میشوند.
همبستگی واضحی بین محل جهش و فنوتیپ وجود دارد1).
| محل جهش | فنوتیپ مرتبط |
|---|---|
| کدونهای 311 تا 1444 | CHRPE |
| کدون 1578-767 | استئوما |
| اگزون 1399 به بعد | دسموئید (خطر 800 برابر) |
| کدون 1309 | سرطان روده بزرگ زودرس در سن 20 سالگی |
ناحیه خوشه جهش (MCR) شامل کدونهای 1250 تا 1464 است 1)، شایعترین جهش در کدون 1309 (حدود 10٪ از کل) و پس از آن کدون 1061 (حدود 5٪) است 1). سن شروع سرطان کولورکتال نیز با توجه به محل جهش متفاوت است: برای کدون 1309 حدود 20 سال، برای کدونهای 168 تا 1580 حدود 30 سال، و برای سایر موارد حدود 52 سال تخمین زده میشود 1).
آزمایش مولکولی ژن APC از دوران کودکی قابل انجام است. کولونوسکوپی معمولاً از حدود 10 سالگی توصیه میشود. در صورت شناسایی جهش APC در خانواده، تأیید وجود یا عدم وجود جهش با آزمایش ژنتیک مهم است.
برای تشخیص قطعی، آزمایش ژنتیک مولکولی ژن APC انجام میشود. آزمایش پانل توالییابی نسل بعدی (NGS) توصیه میگردد 1).
آزمایش مولکولی ژن APC مبنای تشخیص قطعی GS است، اما در برخی بیماران جهش شناسایی نمیشود. استفاده از پانلهای NGS میزان تشخیص را بهبود بخشیده است1). نتایج آزمایش ژنتیکی باید در ترکیب با یافتههای بالینی به طور جامع ارزیابی شود.
CHRPE به خودی خود بدخیم نمیشود و تأثیری بر عملکرد بینایی ندارد. مداخله چشمی لازم نیست و فقط پیگیری دورهای انجام میشود.
برای مهار رشد پولیپها، قبل از جراحی یا در موارد خفیف استفاده میشود.
درمان دارویی به تنهایی نمیتواند خطر سرطان روده بزرگ را از بین ببرد.
توصیه میشود کولکتومی تا سن ۲۵ سالگی انجام شود و ideally در سن ۱۶ تا ۲۰ سالگی انجام شود2). سه نوع اصلی جراحی وجود دارد2).
کولکتومی کامل همراه با ایلئوستومی دائمی
ویژگی: مطمئنترین روش جراحی. هیچ اپیتلیوم روده بزرگ باقی نمیماند
مزیت: حداکثر کاهش خطر سرطان روده بزرگ
عیب: نیاز به استومی دائمی دارد و بر کیفیت زندگی تأثیر میگذارد
کولکتومی کامل رکتوم و آناستوموز کیسه ایلئال-مقعدی
ویژگی: روشی که تداوم روده را حفظ میکند
مزیت: از استومی دائمی جلوگیری میکند
عیب: خطر عوارض ادراری و جنسی وجود دارد
کولکتومی کامل و آناستوموز ایلئورکتال
ویژگیها: تهاجم کم، مناسب برای بیماران جوان
مزایا: تهاجم جراحی نسبتاً کم
معایب: نیاز به نظارت مداوم بر رکتوم باقیمانده
تومور دسموئید از نظر بافتشناسی خوشخیم است، اما تهاجم موضعی بالا و عود شایع دارد. درمان نیازمند رویکرد چندتخصصی و جامع است4)5).
پس از برداشتن روده بزرگ، نظارت زیر ادامه مییابد2):
بسته به تعداد پولیپها و درجه دیسپلازی متفاوت است. در صورت پولیپهای کم، ممکن است با داروهایی مانند NSAIDs یا مهارکنندههای COX-2 تحت نظر قرار گیرند. اما در صورت وجود بیش از ۲۰ پولیپ یا پولیپ با دیسپلازی بالا، برداشتن پیشگیرانه توصیه میشود. زمان ایدهآل جراحی ۱۶ تا ۲۰ سالگی ذکر شده است 2).
پروتئین APC یک مهارکننده تومور متشکل از 2800 اسید آمینه است. ساختار دامنه اصلی آن شامل ناحیه الیگومریزاسیون، تکرارهای آرمادیلو، ناحیه اتصال β-کاتنین، ناحیه اتصال آکسین و ناحیه اتصال میکروتوبول است 1).
جهش APC باعث فعالسازی دائمی مسیر سیگنالینگ Wnt میشود. پروتئین APC طبیعی β-کاتنین را فسفریله کرده و تجزیه آن را در پروتئازوم تسریع میکند. در جهش APC، این عملکرد از دست میرود، β-کاتنین در سیتوپلاسم تجمع یافته و در هسته با فاکتور رونویسی TCF/LEF کمپلکس تشکیل داده و رونویسی ژنهای تکثیر سلولی را افزایش میدهد.
بیشتر جهشها از نوع تغییر چارچوب یا بیمعنی هستند که منجر به تولید پروتئین APC کوتاهشده (از دست دادن عملکرد) میشوند 1).
در تومورهای دسموئید پراکنده، جهش در CTNNB1 (ژن β-کاتنین) در بیش از 75% موارد یافت میشود که مسیر مشابهی را درگیر میکند 3).
CHRPE به عنوان یک ناهنجاری تکثیر سلولی موضعی در اپیتلیوم رنگدانهدار شبکیه ایجاد میشود. نواحی تکثیر غیرطبیعی اپیتلیوم رنگدانهدار شبکیه باعث تغلیظ و بزرگشدن رنگدانه شده و در تضاد با لاکونای اطراف (نواحی دپیگمانته) ظاهری مشخص ایجاد میکند. پتانسیل بدخیمی تقریباً وجود ندارد و تأثیری بر عملکرد بینایی ندارد.
آزمایش پانل توالییابی نسل بعد (NGS) دقت شناسایی محل جهشهای ژن APC را بهبود بخشیده است 1). طبقهبندی خطر بر اساس ارتباط ژنوتیپ و فنوتیپ در حال پیشرفت است و انتظار میرود که استراتژیهای غربالگری و درمانی شخصیسازی شده بر اساس محل جهش ایجاد شود 1).
Litchinko و همکاران (2022) یک مورد نادر از تومور دسموئید بزرگ (170 میلیمتر) در پانکراس را گزارش کردند3). مدیریت چندتخصصی توسط تیم چندرشتهای انجام شد و دشواری در تعیین اندیکاسیون و زمان جراحی در دسموئید پانکراس را نشان داد. اثربخشی سورافنیب به عنوان درمان دارویی مورد توجه قرار گرفته و استفاده از آن در مواردی که برداشتن جراحی دشوار است، در حال بررسی است.
کاربرد اولتراسوند متمرکز با شدت بالا (HIFU) به عنوان یک درمان غیرتهاجمی برای دسموئید نیز در مرحله تحقیقاتی است3).
Albuquerque و همکاران (2025) یک مورد از تومور دسموئید کودکان را گزارش کردند که نیاز به بازسازی عملکرد مفصل شانه داشت5). رویکرد چندتخصصی شامل شیمیدرمانی قبل از عمل با وینبلاستین و متوترکسات، حفظ عملکرد و کنترل تومور را به همراه داشت. دسموئیدهای کمربند شانه و دیواره قفسه سینه 37 تا 50 درصد موارد را تشکیل میدهند و اهمیت بازسازی عملکرد در کودکان را نشان میدهند.
Diaz و همکاران (2025) یک مورد از سندرم گاردنر با معضل جراحی شامل کارسینوم سلول بازال مهاجم و تعویض کامل مفصل زانو را گزارش کردند و نیاز به مدیریت چندتخصصی برای عوارض نادر را نشان دادند4). با شیوع 1 در 100,000 تا 160,000 نفر، اهمیت پزشکی فردیشده در این بیماری رو به افزایش است.