Osteom
Häufigkeit: 60–80 % der FAP-Patienten
Prädilektionsstellen: Kieferknochen, Nasennebenhöhlen, Schädelknochen
Diagnostische Bedeutung: 3 oder mehr deuten stark auf GS hin. Kann der FAP-Diagnose um 17 Jahre vorausgehen

Das Gardner-Syndrom (GS) ist eine phänotypische Variante der familiären adenomatösen Polyposis (FAP). Neben der adenomatösen Polyposis coli sind extrakolonische Manifestationen wie Osteome, Weichteiltumore (Desmoidtumore) und epidermale Zysten charakteristisch. Die Vererbung erfolgt autosomal-dominant.
Das verantwortliche Gen ist das APC-Gen (Adenomatous Polyposis Coli), das auf Chromosom 5q21 lokalisiert ist. Die Erkrankung wird durch Keimbahnmutationen verursacht, und die Vererbungswahrscheinlichkeit an die Nachkommen beträgt 50%. Die Prävalenz wird auf 1 pro 100.000 bis 160.000 Personen geschätzt4).
FAP hat die folgenden Subtypen 1).
FAP macht etwa 1 % aller kolorektalen Karzinome aus 1). Kolonpolypen treten ab etwa dem 10. Lebensjahr auf und können mehrere Hundert bis Zehntausend betragen. Unbehandelt entwickeln 50 % der Patienten bis zum 40. Lebensjahr und nahezu alle bis zum 60. Lebensjahr ein kolorektales Karzinom. De-novo-Mutationen treten bei etwa 20–30 % der Fälle auf 1)5).
Da De-novo-Mutationen im APC-Gen mit einer Häufigkeit von etwa 20–30 % auftreten, kann die Erkrankung auch ohne familiäre Vorbelastung entstehen 1)5). Es ist wichtig zu beachten, dass das Fehlen einer Familienanamnese das Gardner-Syndrom nicht ausschließt.
Darmpolypen sind in der Regel asymptomatisch und werden oft spät entdeckt. Wenn Symptome auftreten, stehen folgende im Vordergrund.
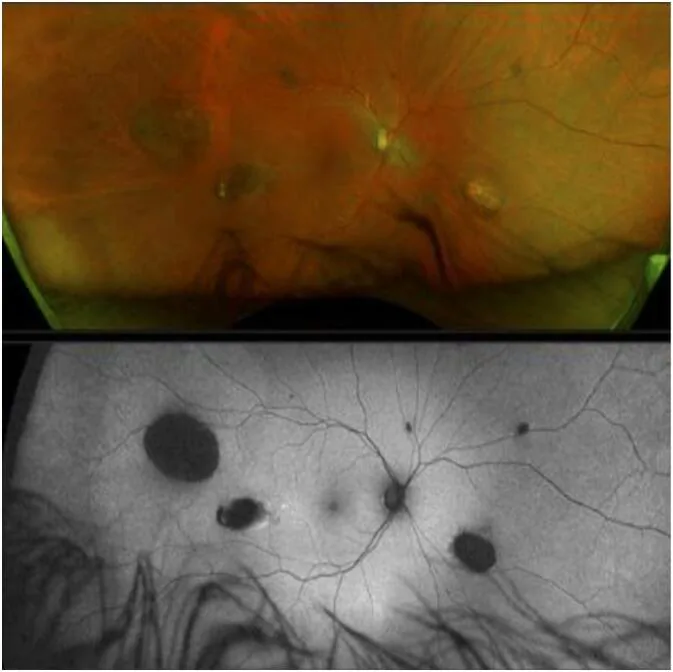
CHRPE (congenital hypertrophy of the retinal pigment epithelium) ist das früheste extrakolonische Zeichen der GS und besteht seit der Geburt 1).
Allerdings kann GS auch ohne Nachweis einer CHRPE nicht ausgeschlossen werden2).
Osteom
Häufigkeit: 60–80 % der FAP-Patienten
Prädilektionsstellen: Kieferknochen, Nasennebenhöhlen, Schädelknochen
Diagnostische Bedeutung: 3 oder mehr deuten stark auf GS hin. Kann der FAP-Diagnose um 17 Jahre vorausgehen
CHRPE
Häufigkeit: 74% der FAP-Patienten
Merkmale: beidseitig, multipel, erbsengroß. Das früheste Anzeichen, das von Geburt an vorhanden ist
Hinweis: GS kann auch ohne CHRPE nicht ausgeschlossen werden
Epidermalzyste
Häufigkeit: ca. 70%
Prädilektionsstelle: Kopf-Hals-Bereich
Merkmale: Tritt bereits in jungen Jahren auf und ist oft multipel vorhanden
Desmoidtumor
Häufigkeit: 12–15 % der FAP-Patienten
Prädilektionsstellen: Abdomen (37–50 %), gefolgt von Schultergürtel/Brustwand
Hinweis: Zweithäufigste Todesursache bei FAP-Patienten nach kolorektalem Karzinom. Mortalität 10–50%
Bei 30–75% der FAP-Patienten feststellbar 1). Retinierte Zähne und überzählige Zähne (bei 11–27% der FAP-Patienten gegenüber 0–4% in der Allgemeinbevölkerung) treten auf 1).
Auch außerhalb des Dickdarms steigt das Risiko für verschiedene bösartige Tumore.
| Krebsart | Lebenszeitrisiko |
|---|---|
| Schilddrüsenkrebs | FAP 2–12 %, Gardner-Typ 10 % |
| Dünndarmkrebs | 4–12 % |
| Bauchspeicheldrüsenkrebs | etwa 1 % |
(Quelle: Zitat 1) 1)
Da eine isolierte CHRPE auch in der Allgemeinbevölkerung mit einer Häufigkeit von 1,2–4,4 % auftritt, kann allein aufgrund des Vorhandenseins von CHRPE nicht mit Sicherheit auf ein GS geschlossen werden. GS-assoziierte CHRPE sind typischerweise beidseitig, multipel, erbsenförmig und unscharf begrenzt. Umgekehrt kann ein GS auch ohne Nachweis von CHRPE nicht ausgeschlossen werden 2).
Die Ursache des GS ist eine Keimbahnmutation im APC-Gen (5q21). Die meisten Mutationen sind Frameshift- oder Nonsense-Mutationen, die zur Produktion eines verkürzten (funktionsverlust) APC-Proteins führen.
Es besteht eine klare Korrelation zwischen der Mutationsstelle und dem Phänotyp 1).
| Mutationsstelle | Zugehöriger Phänotyp |
|---|---|
| Codon 311–1444 | CHRPE |
| Codon 767–1578 | Osteom |
| Exon 1399 und später | Desmoid (800-faches Risiko) |
| Codon 1309 | Frühzeitiges kolorektales Karzinom im Alter von 20 Jahren |
Die Mutationsclusterregion (MCR) umfasst die Codons 1250–1464 1), wobei die häufigste Mutation Codon 1309 (etwa 10 % aller Fälle) und die zweithäufigste Codon 1061 (etwa 5 %) ist 1). Das Erkrankungsalter für Darmkrebs variiert je nach Mutationsort: Bei Codon 1309 liegt es bei etwa 20 Jahren, bei Codon 168–1580 bei etwa 30 Jahren und bei anderen Mutationen bei etwa 52 Jahren 1).
Molekulargenetische Tests des APC-Gens können bereits im Kindesalter durchgeführt werden. Eine Darmspiegelung wird oft ab etwa 10 Jahren empfohlen. Wenn in der Familie eine APC-Mutation bekannt ist, ist es wichtig, das Vorhandensein der Mutation durch einen Gentest zu bestätigen.
Die definitive Diagnose erfolgt durch molekulargenetische Tests des APC-Gens. Ein Next-Generation-Sequencing (NGS)-Panel-Test wird empfohlen 1).
Die molekulargenetische Untersuchung des APC-Gens liefert den Nachweis für die definitive Diagnose des GS, jedoch wird bei einigen Patienten keine Mutation nachgewiesen. Durch den Einsatz von NGS-Panel-Tests hat sich die Nachweisrate verbessert 1). Die Ergebnisse der Gentests werden in Kombination mit den klinischen Befunden umfassend beurteilt.
CHRPE selbst wird nicht bösartig und hat keine Auswirkungen auf die Sehfunktion. Ein augenärztlicher Eingriff ist nicht erforderlich; es erfolgt lediglich eine regelmäßige Beobachtung.
Zur Hemmung des Polypenwachstums wird sie vor Operationen oder bei leichten Fällen eingesetzt.
Eine alleinige medikamentöse Therapie kann das Risiko für Darmkrebs nicht ausschließen.
Die Kolektomie wird bis zum Alter von 25 Jahren empfohlen, idealerweise zwischen 16 und 20 Jahren 2). Die wichtigsten Operationsverfahren sind die folgenden drei 2).
Totale Proktokolektomie mit permanentem Ileostoma
Merkmal: Das sicherste Verfahren. Es verbleibt kein Dickdarmepithel.
Vorteil: Maximales Risiko für Dickdarmkrebs wird reduziert.
Nachteile: Ein permanentes Stoma ist erforderlich und beeinträchtigt die Lebensqualität
Proktokolektomie mit ileoanaler Pouchanastomose
Merkmale: Operationsverfahren, das die Darmkontinuität erhält
Vorteile: Ein permanentes Stoma kann vermieden werden
Nachteile: Risiko von urologischen und sexuellen Funktionsstörungen
Totale Kolektomie mit ileorektaler Anastomose
Merkmale: Weniger invasiv, geeignet für junge Patienten
Vorteile: Relativ geringer chirurgischer Eingriff
Nachteile: Eine kontinuierliche Überwachung des verbleibenden Rektums ist erforderlich
Desmoidtumoren sind histologisch gutartig, aber lokal invasiv und neigen zu Rezidiven. Die Behandlung erfordert einen multidisziplinären Ansatz4)5).
Auch nach einer Kolonresektion wird die folgende Überwachung fortgesetzt2).
Das hängt von der Anzahl der Polypen und dem Grad der Dysplasie ab. Bei wenigen Polypen kann eine medikamentöse Therapie mit NSAIDs oder COX-2-Hemmern erwogen werden. Bei 20 oder mehr Polypen oder hochgradiger Dysplasie wird jedoch eine prophylaktische Resektion empfohlen. Der ideale Operationszeitpunkt liegt zwischen 16 und 20 Jahren 2).
Das APC-Protein ist ein Tumorsuppressor, der aus 2800 Aminosäuren besteht. Zu den wichtigsten Domänenstrukturen gehören eine Oligomerisierungsregion, Armadillo-Repeats, eine β-Catenin-Bindungsregion, eine Axin-Bindungsregion und eine Mikrotubuli-Bindungsregion 1).
APC-Mutationen führen zu einer konstitutiven Aktivierung des Wnt-Signalwegs. Normales APC-Protein phosphoryliert β-Catenin und fördert dessen Abbau im Proteasom. Bei APC-Mutationen geht diese Funktion verloren, β-Catenin akkumuliert im Zytoplasma, bildet im Zellkern einen Komplex mit TCF/LEF-Transkriptionsfaktoren und fördert die Transkription von Zellproliferationsgenen.
Die meisten Mutationen sind Frameshift- oder Nonsense-Mutationen, die zu einem verkürzten (funktionsverlust) APC-Protein führen 1).
Bei sporadischen Desmoidtumoren werden in über 75% der Fälle Mutationen im CTNNB1 (β-Catenin-Gen) gefunden, was auf einen ähnlichen Signalweg hindeutet 3).
CHRPE entsteht als lokale Zellproliferationsanomalie des retinalen Pigmentepithels. In den Bereichen mit abnormaler Proliferation des retinalen Pigmentepithels kommt es zu einer Konzentration und Hypertrophie des Pigments, was im Kontrast zu den umgebenden Lakunen (depigmentierten Bereichen) ein charakteristisches Erscheinungsbild ergibt. Es besteht nahezu kein malignes Potenzial, und es werden keine Auswirkungen auf die Sehfunktion festgestellt.
Durch Next-Generation-Sequencing (NGS)-Panel-Tests hat sich die Genauigkeit der Identifizierung von Mutationen im APC-Gen verbessert 1). Die Risikostratifizierung auf der Grundlage von Genotyp-Phänotyp-Korrelationen schreitet voran, und es wird erwartet, dass personalisierte Überwachungs- und Behandlungsstrategien entsprechend der Mutationsstelle entwickelt werden 1).
Litchinko et al. (2022) berichteten über einen seltenen Fall eines großen Desmoidtumors (170 mm) der Bauchspeicheldrüse3). Ein multidisziplinäres Team führte ein umfassendes Management durch und zeigte die Schwierigkeit der Operationsindikation und des Zeitpunkts bei Pankreasdesmoiden auf. Als medikamentöse Therapie wird die Wirksamkeit von Sorafenib hervorgehoben, und sein Einsatz bei Fällen, bei denen eine chirurgische Resektion schwierig ist, wird untersucht.
Die Anwendung des hochintensiven fokussierten Ultraschalls (HIFU) zur nicht-invasiven Behandlung von Desmoiden befindet sich ebenfalls in der Forschungsphase3).
Albuquerque et al. (2025) berichteten über einen Fall eines pädiatrischen Desmoidtumors, der eine Rekonstruktion der Schultergelenksfunktion erforderte5). Durch einen multidisziplinären Ansatz einschließlich präoperativer Chemotherapie mit Vinblastin und Methotrexat wurden Funktionserhalt und Tumorkontrolle erreicht. Desmoide des Schultergürtels und der Brustwand machen 37–50 % aller Fälle aus, was die Bedeutung der Funktionsrekonstruktion bei Kindern unterstreicht.
Diaz et al. (2025) berichteten über GS-Fälle mit chirurgischen Dilemmata, einschließlich invasivem Basalzellkarzinom und Knie-Totalendoprothese, und zeigten, dass ein multidisziplinäres Management seltener Komplikationen erforderlich ist 4). Bei dieser Erkrankung, deren Prävalenz auf 1 pro 100.000 bis 160.000 Personen geschätzt wird, nimmt die Bedeutung der personalisierten Medizin zu.