Остеома
Частота: 60–80% пациентов с САП
Излюбленная локализация: челюсти, околоносовые пазухи, череп
Диагностическое значение: 3 и более сильно указывают на ГС. Могут предшествовать диагнозу САП на 17 лет

Синдром Гарднера (Gardner syndrome; GS) — один из фенотипических вариантов семейного аденоматозного полипоза (familial adenomatous polyposis; FAP). Характеризуется аденоматозным полипозом толстой кишки в сочетании с внекишечными проявлениями, такими как остеомы, опухоли мягких тканей (десмоидные опухоли) и эпидермальные кисты. Наследуется по аутосомно-доминантному типу.
Ген-кандидат — ген APC (adenomatous polyposis coli), расположенный на длинном плече 5-й хромосомы (5q21). Заболевание вызывается герминативными мутациями, вероятность передачи потомству составляет 50%. Распространенность оценивается как 1 случай на 100 000–160 000 человек4).
FAP имеет следующие подтипы1).
FAP составляет около 1% всех случаев рака толстой кишки1). Полипы толстой кишки появляются примерно в возрасте 10 лет и достигают от нескольких сотен до десятков тысяч. Без лечения к 40 годам рак толстой кишки развивается у 50% пациентов, а к 60 годам — практически у всех. De novo мутации (новые мутации) наблюдаются примерно в 20–30% случаев1)5).
Поскольку de novo мутации (новые мутации) в гене APC возникают с частотой около 20–30%, заболевание может развиться и при отсутствии семейного анамнеза1)5). Важно отметить, что отсутствие семейного анамнеза не позволяет исключить синдром Гарднера.
Полипы толстой кишки обычно протекают бессимптомно, и их обнаружение часто запаздывает. При появлении симптомов преобладают следующие.
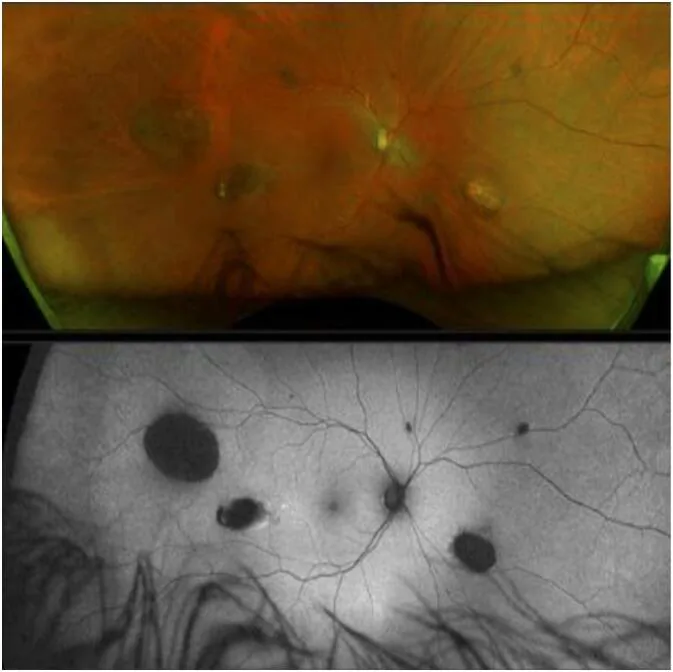
CHRPE (врожденная гипертрофия пигментного эпителия сетчатки) является наиболее ранним внекишечным проявлением GS и присутствует с рождения 1).
Однако отсутствие CHRPE не исключает GS2).
Остеома
Частота: 60–80% пациентов с САП
Излюбленная локализация: челюсти, околоносовые пазухи, череп
Диагностическое значение: 3 и более сильно указывают на ГС. Могут предшествовать диагнозу САП на 17 лет
ХРПЭ
Частота: 74% пациентов с САП
Особенности: двусторонние, множественные, размером с горошину. Самый ранний признак, присутствующий с рождения
Внимание: отсутствие CHRPE не исключает GS
Эпидермальная киста
Частота: около 70%
Излюбленная локализация: голова и шея
Особенности: появляется в молодом возрасте, часто множественные
Десмоидная опухоль
Частота: 12–15% пациентов с FAP
Излюбленная локализация: живот (37–50%), затем плечевой пояс и грудная стенка
Внимание: вторая по частоте причина смерти пациентов с САП после колоректального рака. Смертность 10–50%
Наблюдаются у 30–75% пациентов с САП1). Отмечаются ретенированные и сверхкомплектные зубы (у 11–27% пациентов с САП против 0–4% в общей популяции)1).
Повышается риск различных злокачественных опухолей, помимо колоректального рака.
| Тип рака | Пожизненный риск |
|---|---|
| Рак щитовидной железы | FAP 2–12%, тип Гарднера 10% |
| Рак тонкой кишки | 4–12% |
| Рак поджелудочной железы | около 1% |
(Источник: ссылка 1)1)
Причиной GS является герминальная мутация гена APC (5q21). Большинство мутаций представляют собой сдвиг рамки считывания или нонсенс-мутации, приводящие к продукции усеченного (потерявшего функцию) белка APC.
Существует четкая корреляция между локализацией мутации и фенотипом 1).
| Локализация мутации | Связанный фенотип |
|---|---|
| кодоны 311–1444 | CHRPE |
| кодоны 767–1578 | остеома |
| Экзон 1399 и далее | Десмоид (риск в 800 раз) |
| Кодон 1309 | Рак толстой кишки в возрасте 20 лет |
Область кластера мутаций (mutation cluster region; MCR) находится между кодонами 1250 и 14641), наиболее частой мутацией является кодон 1309 (около 10% всех случаев), затем кодон 1061 (около 5%)1). Возраст начала колоректального рака также различается в зависимости от места мутации: для кодона 1309 — 20 лет, для кодонов 168–1580 — около 30 лет, для остальных — около 52 лет1).
Молекулярно-генетическое тестирование гена APC можно проводить с раннего детства. Колоноскопию часто рекомендуют начинать примерно с 10 лет. Если в семье выявлена мутация APC, важно подтвердить наличие или отсутствие мутации с помощью генетического теста.
Для окончательного диагноза проводится молекулярно-генетическое тестирование гена APC. Рекомендуется панельное тестирование методом секвенирования нового поколения (NGS) 1).
Молекулярно-генетическое исследование гена APC является основанием для подтверждения диагноза ГС, однако у некоторых пациентов мутации могут не обнаруживаться. Использование панельного NGS-тестирования повышает частоту выявления 1). Результаты генетического теста оцениваются в совокупности с клиническими данными.
Сама по себе CHRPE не малигнизируется и не влияет на зрительные функции. Офтальмологического вмешательства не требуется, проводится только регулярное наблюдение.
Для подавления роста полипов применяется перед операцией или при легких случаях.
Медикаментозная терапия сама по себе не может устранить риск колоректального рака.
Колэктомию рекомендуется проводить до 25 лет, в идеале — в возрасте 16–20 лет2). Существуют три основных типа операций2).
Тотальная колэктомия с проктэктомией и постоянной илеостомой
Особенности: наиболее радикальная операция. Толстокишечный эпителий не сохраняется.
Преимущества: максимальное снижение риска рака толстой кишки.
Недостатки: требуется постоянная стома, что влияет на качество жизни
Тотальная проктоколэктомия с восстановительным илеоанальным анастомозом
Особенности: операция, сохраняющая непрерывность кишечника
Преимущества: позволяет избежать постоянной стомы
Недостатки: риск урологических и сексуальных осложнений
Тотальная колэктомия + илеоректальный анастомоз
Особенности: малая инвазивность, подходит для молодых пациентов
Преимущества: относительно малая хирургическая травма
Недостаток: требуется постоянное наблюдение за оставшейся прямой кишкой
Десмоидные опухоли гистологически доброкачественны, но обладают высокой местной инвазивностью и склонны к рецидивам. Лечение требует мультидисциплинарного подхода с участием нескольких специалистов4)5).
После резекции толстой кишки наблюдение продолжается следующим образом2).
Это зависит от количества полипов и степени дисплазии. При небольшом количестве полипов может применяться медикаментозная терапия НПВП или ингибиторами ЦОГ-2 с наблюдением. Однако при наличии 20 и более полипов или полипов с высокой степенью дисплазии рекомендуется профилактическое удаление. Идеальным возрастом для операции считается 16–20 лет 2).
Белок APC является супрессором опухоли, состоящим из 2800 аминокислот. Основные доменные структуры включают область олигомеризации, повторы армадилло, область связывания β-катенина, область связывания аксина и область связывания микротрубочек 1).
Мутации APC вызывают конститутивную активацию сигнального пути Wnt. Нормальный белок APC фосфорилирует β-катенин, способствуя его деградации в протеасоме. При мутации APC эта функция утрачивается, β-катенин накапливается в цитоплазме, образует комплекс с факторами транскрипции TCF/LEF в ядре и стимулирует транскрипцию генов, способствующих клеточной пролиферации.
Большинство мутаций представляют собой сдвиг рамки считывания или нонсенс-мутации, приводящие к продукции усеченного (с потерей функции) белка APC 1).
При спорадических десмоидных опухолях мутации CTNNB1 (гена β-катенина) обнаруживаются более чем в 75% случаев, что указывает на вовлечение аналогичного пути 3).
CHRPE возникает как локальное нарушение пролиферации клеток пигментного эпителия сетчатки. В участках аномального разрастания пигментного эпителия сетчатки пигмент концентрируется и гипертрофируется, создавая характерный вид на фоне окружающих лакун (областей депигментации). Потенциал злокачественного перерастания практически отсутствует, и влияния на зрительную функцию не наблюдается.
Панельное тестирование секвенирования нового поколения (NGS) повышает точность идентификации мутаций в гене APC1). Прогресс в стратификации риска на основе корреляции генотип-фенотип позволяет разрабатывать индивидуальные стратегии наблюдения и лечения в зависимости от локализации мутации1).
Litchinko и соавт. (2022) сообщили о редком случае крупной десмоидной опухоли поджелудочной железы (170 мм)3). Мультидисциплинарное ведение команды специалистов продемонстрировало сложность определения показаний и сроков хирургического вмешательства при десмоиде поджелудочной железы. В качестве лекарственной терапии отмечается эффективность сорафениба, рассматривается его применение в случаях, когда хирургическое удаление затруднено.
Применение высокоинтенсивного фокусированного ультразвука (HIFU) для неинвазивного лечения десмоидов также находится на стадии исследований3).
Albuquerque и соавт. (2025) сообщили о случае детской десмоидной опухоли, потребовавшей реконструкции функции плечевого сустава5). Мультидисциплинарный подход, включающий предоперационную химиотерапию винбластином и метотрексатом, позволил достичь сохранения функции и контроля опухоли. Десмоиды плечевого пояса и грудной стенки составляют 37–50% всех случаев, что подчеркивает важность функциональной реконструкции у детей.
Diaz и соавт. (2025) сообщили о случаях GS с хирургическими дилеммами, включая инвазивную базальноклеточную карциному и тотальное эндопротезирование коленного сустава, что подчеркивает необходимость мультидисциплинарного подхода к редким осложнениям4). При заболеваемости 1 на 100 000–160 000 человек возрастает важность персонализированной медицины.