Osteoma
Frecuencia: 60–80% de los pacientes con FAP
Sitios comunes: Huesos maxilares, senos paranasales, cráneo
Importancia diagnóstica: Tres o más sugieren fuertemente GS. Puede preceder al diagnóstico de FAP por 17 años

El síndrome de Gardner (GS) es una variante fenotípica de la poliposis adenomatosa familiar (PAF). Además de la poliposis adenomatosa colorrectal, se caracteriza por manifestaciones extracolónicas como osteomas, tumores de tejidos blandos (tumores desmoides) y quistes epidérmicos. Sigue un patrón de herencia autosómico dominante.
El gen causante es el gen APC (adenomatous polyposis coli), ubicado en el cromosoma 5q21. Es causado por mutaciones en la línea germinal, con una probabilidad de transmisión a la descendencia del 50%. La prevalencia se estima en 1 de cada 100,000 a 160,000 individuos4).
La FAP tiene los siguientes subtipos 1).
La FAP representa aproximadamente el 1% de todos los cánceres colorrectales 1). Los pólipos colorrectales aparecen alrededor de los 10 años y pueden llegar a cientos o decenas de miles. Sin tratamiento, el 50% desarrolla cáncer colorrectal a los 40 años y casi todos a los 60 años. Las mutaciones de novo se encuentran en aproximadamente el 20–30% de los casos 1)5).
Las mutaciones de novo en el gen APC ocurren con una frecuencia de aproximadamente el 20–30%, por lo que la enfermedad puede presentarse incluso sin antecedentes familiares1)5). Es importante tener en cuenta que la ausencia de antecedentes familiares no descarta el GS.
Los pólipos colorrectales suelen ser asintomáticos y su detección a menudo se retrasa. Cuando aparecen síntomas, principalmente incluyen lo siguiente.
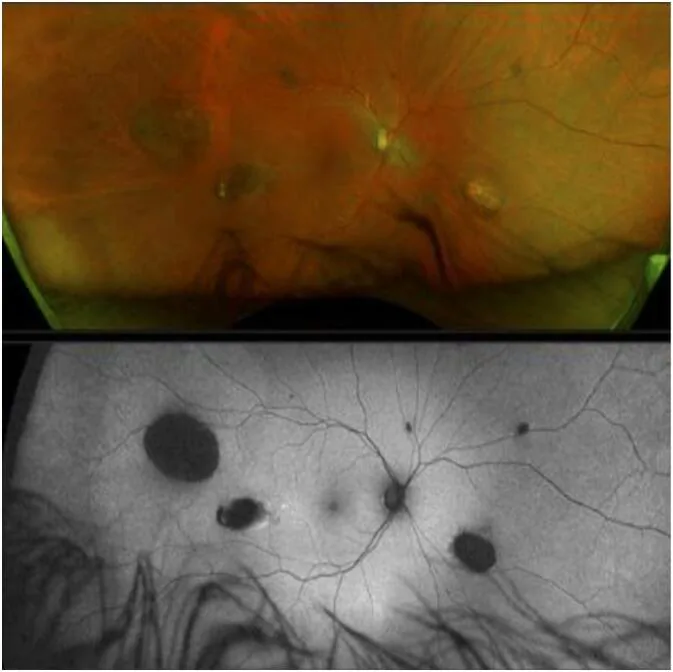
La CHRPE (hipertrofia congénita del epitelio pigmentario de la retina) es la manifestación extracolónica más temprana de GS y está presente desde el nacimiento 1).
Sin embargo, la GS no puede descartarse incluso si no se observa CHRPE 2).
Osteoma
Frecuencia: 60–80% de los pacientes con FAP
Sitios comunes: Huesos maxilares, senos paranasales, cráneo
Importancia diagnóstica: Tres o más sugieren fuertemente GS. Puede preceder al diagnóstico de FAP por 17 años
CHRPE
Frecuencia: 74% de los pacientes con FAP
Características: Bilateral, múltiple, en forma de guisante. El signo más temprano presente desde el nacimiento
Nota: No se puede descartar GS incluso sin CHRPE
Quiste epidérmico
Frecuencia: Aproximadamente 70%
Sitio común: Cabeza y cuello
Características: Aparece desde una edad temprana, a menudo múltiple
Tumor desmoide
Frecuencia: 12–15% de los pacientes con FAP
Sitios comunes: Abdomen (37–50%), seguido de cintura escapular/pared torácica
Nota: Segunda causa de muerte en pacientes con FAP después del cáncer colorrectal. Mortalidad del 10–50%
Se observa en el 30–75% de los pacientes con FAP1). Se presentan dientes impactados y supernumerarios (11–27% en pacientes con FAP frente a 0–4% en la población general)1).
El riesgo de varios tumores malignos distintos del colorrectal también aumenta.
| Tipo de cáncer | Riesgo de por vida |
|---|---|
| Cáncer de tiroides | FAP 2–12%, tipo Gardner 10% |
| Cáncer de intestino delgado | 4–12% |
| Cáncer de páncreas | Aproximadamente 1% |
(Fuente: Referencia 1)1)
La CHRPE esporádica se encuentra en el 1.2–4.4% de la población general, por lo que la presencia de CHRPE por sí sola no confirma GS. La CHRPE asociada a GS se caracteriza por lesiones bilaterales, múltiples, en forma de guisante y con bordes irregulares. Por el contrario, la GS no puede descartarse incluso si no hay CHRPE2).
La causa de GS es una mutación germinal en el gen APC (5q21). La mayoría de las mutaciones son de cambio de marco o sin sentido, lo que produce una proteína APC truncada (sin función).
Existe una clara correlación entre el sitio de la mutación y el fenotipo1).
| Sitio de la mutación | Fenotipo asociado |
|---|---|
| Codones 311–1444 | CHRPE |
| Codones 767–1578 | Osteoma |
| Exón 1399 en adelante | Tumor desmoide (riesgo 800 veces mayor) |
| Codón 1309 | Cáncer colorrectal temprano a los 20 años |
La región de agrupación de mutaciones (MCR) abarca los codones 1250 a 1464 1), siendo la mutación más frecuente en el codón 1309 (aproximadamente el 10% del total), seguida del codón 1061 (aproximadamente el 5%) 1). La edad de aparición del cáncer colorrectal también varía según el sitio de la mutación: alrededor de los 20 años para el codón 1309, alrededor de los 30 años para los codones 168-1580, y alrededor de los 52 años para otros sitios 1).
La prueba genética molecular del gen APC se puede realizar desde la primera infancia. A menudo se recomienda iniciar la colonoscopia alrededor de los 10 años. Si se conoce una mutación de APC en la familia, es importante confirmar la presencia o ausencia de la mutación mediante pruebas genéticas.
Para el diagnóstico definitivo se realiza una prueba genética molecular del gen APC. Se recomienda la prueba de panel de secuenciación de nueva generación (NGS) 1).
La prueba genética molecular del gen APC proporciona la base para el diagnóstico confirmatorio de GS, pero en algunos pacientes no se detectan mutaciones. La tasa de detección ha mejorado con el uso de paneles NGS 1). Los resultados de las pruebas genéticas deben interpretarse en combinación con los hallazgos clínicos.
La CHRPE en sí misma no se vuelve maligna y no afecta la función visual. No es necesaria una intervención oftalmológica, solo se realiza una observación periódica.
Se utiliza antes de la cirugía o en casos leves para suprimir el crecimiento de pólipos.
La medicación por sí sola no puede eliminar el riesgo de cáncer colorrectal.
Se recomienda realizar la colectomía antes de los 25 años, idealmente entre los 16 y 20 años2). Los principales procedimientos quirúrgicos son los siguientes tres tipos2).
Proctocolectomía Total con Ileostomía Permanente
Características: El procedimiento más definitivo. No queda epitelio colorrectal.
Ventajas: Reduce al máximo el riesgo de cáncer colorrectal.
Desventajas: Requiere un estoma permanente, afectando la calidad de vida
Proctocolectomía Restauradora con Anastomosis de Bolsa Ileal-Anal
Características: Procedimiento quirúrgico que preserva la continuidad intestinal
Ventajas: Evita un estoma permanente
Desventajas: Riesgo de complicaciones urinarias y de la función sexual
Colectomía Total con Anastomosis Ileorrectal
Características: Mínimamente invasivo, adecuado para pacientes jóvenes
Ventajas: Relativamente menos invasivo quirúrgicamente
Desventajas: Se requiere vigilancia continua del recto remanente
Los tumores desmoides son histológicamente benignos, pero altamente invasivos localmente y propensos a recurrencia. El tratamiento requiere un enfoque multidisciplinario 4)5).
Continuar la siguiente vigilancia incluso después de la colectomía2).
Depende del número de pólipos y del grado de displasia. Si hay pocos pólipos, se puede optar por tratamiento farmacológico con AINEs o inhibidores de la COX-2 y observación. Sin embargo, si se encuentran 20 o más pólipos o pólipos con displasia de alto grado, se recomienda la resección profiláctica. La edad ideal para la cirugía se considera entre los 16 y 20 años 2).
La proteína APC es un supresor tumoral de 2800 aminoácidos. Sus principales dominios estructurales incluyen una región de oligomerización, repeticiones armadillo, una región de unión a β-catenina, una región de unión a axina y una región de unión a microtúbulos 1).
Las mutaciones de APC causan activación constitutiva de la vía de señalización Wnt. La proteína APC normal fosforila la β-catenina, promoviendo su degradación por el proteasoma. En las mutaciones de APC, esta función se pierde, lo que lleva a la acumulación de β-catenina en el citoplasma, que forma un complejo con factores de transcripción TCF/LEF en el núcleo y promueve la transcripción de genes de proliferación celular.
La mayoría de las mutaciones son de cambio de marco o sin sentido, lo que produce una proteína APC truncada (con pérdida de función) 1).
En los tumores desmoides esporádicos, se encuentran mutaciones de CTNNB1 (gen de la β-catenina) en más del 75% de los casos, involucrando una vía similar 3).
La CHRPE surge como una proliferación anormal localizada del epitelio pigmentario de la retina. Las áreas de proliferación anormal del epitelio pigmentario retiniano muestran pigmento concentrado e hipertrofiado, creando una apariencia característica en contraste con las lagunas circundantes (áreas despigmentadas). Casi no tiene potencial maligno ni afecta la función visual.
Las pruebas de panel de secuenciación de próxima generación (NGS) han mejorado la precisión en la identificación de sitios de mutación en el gen APC 1). La estratificación de riesgo basada en correlaciones genotipo-fenotipo está avanzando, y se espera el desarrollo de estrategias de vigilancia y tratamiento personalizadas según el sitio de mutación 1).
Litchinko et al. (2022) reportaron un caso raro de un tumor desmoide grande (170 mm) en el páncreas 3). Se realizó un manejo multidisciplinario, destacando la dificultad de la indicación y el momento quirúrgico en los desmoides pancreáticos. La eficacia de sorafenib como farmacoterapia está atrayendo la atención, y se está considerando su aplicación en casos donde la resección quirúrgica es difícil.
La aplicación de ultrasonido focalizado de alta intensidad (HIFU) como tratamiento no invasivo para desmoides también se encuentra en fase de investigación 3).
Albuquerque et al. (2025) reportaron un caso de tumor desmoide pediátrico que requirió reconstrucción funcional de la articulación del hombro 5). Un enfoque multidisciplinario que incluyó quimioterapia preoperatoria con vinblastina más metotrexato logró la preservación funcional y el control tumoral. Los desmoides de la cintura escapular y la pared torácica representan el 37–50% de los casos, lo que indica la importancia de la reconstrucción funcional en pacientes pediátricos.
Diaz et al. (2025) reportaron un caso de GS que presentaba dilemas quirúrgicos, incluyendo carcinoma basocelular infiltrante y complicaciones de artroplastia total de rodilla, demostrando la necesidad de un manejo multidisciplinario de complicaciones raras 4). Con una prevalencia de 1 en 100,000–160,000, la importancia de la medicina personalizada está aumentando.