Osteoma
Frequency: 60–80% of FAP patients
Common sites: Jaw bones, paranasal sinuses, skull
Diagnostic significance: Three or more strongly suggest GS. May precede FAP diagnosis by 17 years

Gardner syndrome (GS) is a phenotypic variant of familial adenomatous polyposis (FAP). In addition to colorectal adenomatous polyposis, it is characterized by extracolonic manifestations such as osteomas, soft tissue tumors (desmoid tumors), and epidermal cysts. It follows an autosomal dominant inheritance pattern.
The causative gene is the APC (adenomatous polyposis coli) gene, located on chromosome 5q21. It is caused by germline mutations, with a 50% probability of transmission to offspring. The prevalence is estimated at 1 in 100,000 to 160,000 individuals4).
FAP has the following subtypes 1).
FAP accounts for about 1% of all colorectal cancers 1). Colorectal polyps appear around age 10 and can number from hundreds to tens of thousands. Without treatment, 50% develop colorectal cancer by age 40, and nearly all by age 60. De novo mutations are found in about 20–30% of cases 1)5).
De novo mutations in the APC gene occur with a frequency of approximately 20–30%, so the condition can develop even without a family history1)5). It is important to note that the absence of a family history does not rule out GS.
Colorectal polyps are usually asymptomatic, and detection is often delayed. When symptoms do appear, they mainly include the following.
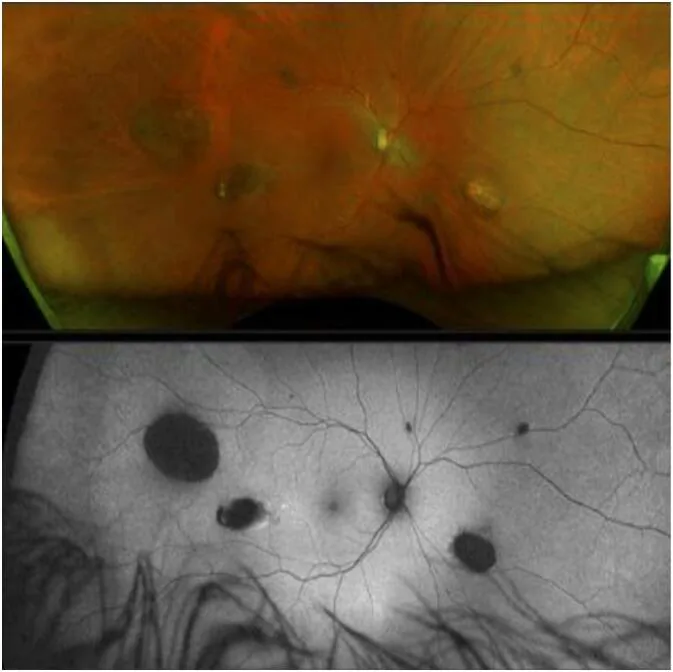
CHRPE (congenital hypertrophy of the retinal pigment epithelium) is the earliest extracolonic manifestation of GS and is present from birth 1).
However, GS cannot be ruled out even if CHRPE is not present 2).
Osteoma
Frequency: 60–80% of FAP patients
Common sites: Jaw bones, paranasal sinuses, skull
Diagnostic significance: Three or more strongly suggest GS. May precede FAP diagnosis by 17 years
CHRPE
Frequency: 74% of FAP patients
Features: Bilateral, multiple, pea-shaped. The earliest sign present from birth
Note: GS cannot be ruled out even without CHRPE
Epidermal cyst
Frequency: Approximately 70%
Common site: Head and neck
Characteristics: Appears from a young age, often multiple
Desmoid tumor
Frequency: 12–15% of FAP patients
Common sites: Abdomen (37–50%), followed by shoulder girdle/chest wall
Note: Second only to colorectal cancer as a cause of death in FAP patients. Mortality rate 10–50%
Found in 30–75% of FAP patients1). Impacted teeth and supernumerary teeth (11–27% in FAP patients vs. 0–4% in the general population) are observed1).
The risk of various malignant tumors other than colorectal cancer also increases.
| Cancer type | Lifetime risk |
|---|---|
| Thyroid cancer | FAP 2–12%, Gardner type 10% |
| Small bowel cancer | 4–12% |
| Pancreatic cancer | Approximately 1% |
(Source: Reference 1)1)
The cause of GS is a germline mutation in the APC gene (5q21). Most mutations are frameshift or nonsense mutations, resulting in a truncated (loss-of-function) APC protein.
There is a clear correlation between mutation site and phenotype1).
| Mutation site | Associated phenotype |
|---|---|
| Codons 311–1444 | CHRPE |
| Codons 767–1578 | Osteoma |
| Exon 1399 and beyond | Desmoid (800-fold risk) |
| Codon 1309 | Early colorectal cancer at age 20 |
The mutation cluster region (MCR) spans codons 1250 to 1464 1), with the most common mutation at codon 1309 (approximately 10% of all cases), followed by codon 1061 (approximately 5%) 1). The age of onset of colorectal cancer also varies by mutation site: around age 20 for codon 1309, around age 30 for codons 168–1580, and around age 52 for other sites 1).
Molecular genetic testing of the APC gene can be performed from early childhood. Colonoscopy is often recommended starting around age 10. If an APC mutation is known in the family, it is important to confirm the presence or absence of the mutation through genetic testing.
For a definitive diagnosis, molecular genetic testing of the APC gene is performed. Next-generation sequencing (NGS) panel testing is recommended 1).
Molecular genetic testing of the APC gene provides a basis for confirming the diagnosis of GS, but mutations may not be detected in some patients. The detection rate has improved with the use of NGS panel testing 1). Genetic test results should be interpreted in combination with clinical findings.
CHRPE itself does not become malignant and has no effect on visual function. Ophthalmic intervention is unnecessary, and only periodic observation is performed.
It is used before surgery or in mild cases to suppress polyp growth.
Medication alone cannot eliminate the risk of colorectal cancer.
Colectomy is recommended by age 25, ideally performed between 16 and 20 years of age2). The main surgical procedures are the following three types2).
Total Proctocolectomy with Permanent Ileostomy
Features: The most definitive procedure. No colorectal epithelium remains.
Advantages: Maximally reduces the risk of colorectal cancer.
Disadvantages: Requires a permanent stoma, affecting quality of life
Restorative Proctocolectomy with Ileal Pouch-Anal Anastomosis
Features: Surgical procedure that preserves intestinal continuity
Advantages: Avoids a permanent stoma
Disadvantages: Risk of urinary and sexual function complications
Total Colectomy with Ileorectal Anastomosis
Features: Minimally invasive, suitable for young patients
Advantages: Relatively less surgical invasiveness
Disadvantages: Continuous surveillance of the remaining rectum is required
Desmoid tumors are histologically benign but highly locally invasive and prone to recurrence. Treatment requires a multidisciplinary approach 4)5).
Continue the following surveillance even after colectomy2).
It depends on the number of polyps and the degree of dysplasia. For a small number of polyps, medication with NSAIDs or COX-2 inhibitors may be used for observation. However, if 20 or more polyps or high-grade dysplastic polyps are found, prophylactic resection is recommended. The ideal timing for surgery is considered to be between 16 and 20 years of age 2).
APC protein is a tumor suppressor consisting of 2800 amino acids. Its main domain structures include an oligomerization region, armadillo repeats, a β-catenin binding region, an axin binding region, and a microtubule binding region 1).
APC mutations cause constitutive activation of the Wnt signaling pathway. Normal APC protein phosphorylates β-catenin, promoting its degradation by the proteasome. In APC mutations, this function is lost, leading to accumulation of β-catenin in the cytoplasm, which forms a complex with TCF/LEF transcription factors in the nucleus and promotes transcription of cell proliferation genes.
Most mutations are frameshift or nonsense mutations, resulting in truncated (loss-of-function) APC protein 1).
In sporadic desmoid tumors, CTNNB1 (β-catenin gene) mutations are found in over 75% of cases, involving a similar pathway 3).
CHRPE arises as a localized abnormal proliferation of retinal pigment epithelium. The areas of abnormal retinal pigment epithelium proliferation show concentrated and hypertrophied pigment, creating a characteristic appearance in contrast to the surrounding lacunae (depigmented areas). There is almost no malignant potential and no effect on visual function.
Next-generation sequencing (NGS) panel testing has improved the accuracy of identifying mutation sites in the APC gene 1). Risk stratification based on genotype-phenotype correlations is advancing, and the development of personalized surveillance and treatment strategies according to mutation sites is expected 1).
Litchinko et al. (2022) reported a rare case of a large desmoid tumor (170 mm) arising in the pancreas 3). Multidisciplinary management was performed, highlighting the difficulty of surgical indication and timing in pancreatic desmoids. The efficacy of sorafenib as pharmacotherapy is attracting attention, and its application to cases where surgical resection is difficult is being considered.
The application of high-intensity focused ultrasound (HIFU) as a non-invasive treatment for desmoids is also under investigation 3).
Albuquerque et al. (2025) reported a case of pediatric desmoid tumor requiring shoulder joint functional reconstruction 5). A multidisciplinary approach including preoperative chemotherapy with vinblastine plus methotrexate achieved functional preservation and tumor control. Desmoids of the shoulder girdle and chest wall account for 37–50% of cases, indicating the importance of functional reconstruction in pediatric patients.
Diaz et al. (2025) reported a GS case presenting surgical dilemmas including invasive basal cell carcinoma and total knee arthroplasty complications, demonstrating the need for multidisciplinary management of rare complications 4). With a prevalence of 1 in 100,000–160,000, the importance of personalized medicine is increasing.