Osteom
Sıklık: FAP hastalarının %60-80’i
Sık görülen yerler: Çene kemiği, sinüsler, kafatası
Tanısal önemi: Üç veya daha fazlası GS’yi güçlü bir şekilde düşündürür. FAP tanısından 17 yıl önce ortaya çıkabilir

Gardner sendromu (Gardner syndrome; GS), ailesel adenomatöz polipozis (familial adenomatous polyposis; FAP)‘in bir fenotipik varyantıdır. Kolon adenomatöz polipozisine ek olarak, osteom, yumuşak doku tümörleri (desmoid tümör) ve epidermal kist gibi kolon dışı bulgularla karakterizedir. Otozomal dominant (eski adıyla otozomal dominant) kalıtım gösterir.
Sorumlu gen, 5. kromozomun 5q21 bölgesinde yer alan APC (adenomatous polyposis coli) genidir. Germ hattı mutasyonu sonucu ortaya çıkar ve çocuğa geçme olasılığı %50’dir. Görülme sıklığı 100.000-160.000’de 1 olarak bildirilmiştir4).
FAP aşağıdaki alt tiplere sahiptir 1).
FAP, tüm kolon kanserlerinin yaklaşık %1’ini oluşturur 1). Kolon polipleri yaklaşık 10 yaşından itibaren ortaya çıkar ve yüzlerce ila on binlerce sayıya ulaşır. Tedavi edilmezse 40 yaşında %50, 60 yaşında hemen hemen tüm vakalarda kolon kanseri gelişir. De novo mutasyon (yeni mutasyon) yaklaşık %20-30 oranında görülür 1)5).
APC geninde yaklaşık %20-30 oranında de novo mutasyon (yeni mutasyon) meydana geldiğinden, aile öyküsü olmasa bile hastalık ortaya çıkabilir1)5). Aile öyküsü olmamasının GS’yi dışlamadığına dikkat edilmelidir.
Kolorektal polipler genellikle asemptomatiktir ve sıklıkla geç teşhis edilir. Semptomlar ortaya çıktığında aşağıdakiler baskındır.
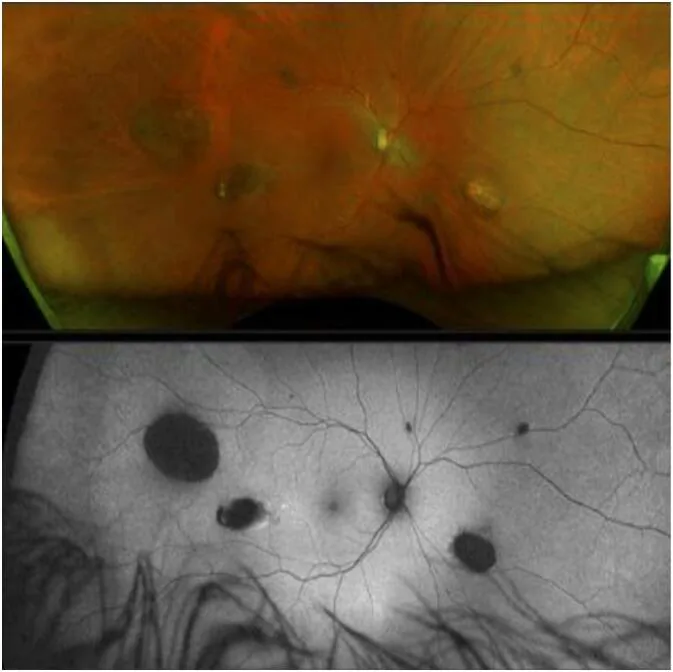
CHRPE (retina pigment epitelinin konjenital hipertrofisi), GS’nin en erken ekstrakolonik bulgusudur ve doğumdan itibaren mevcuttur 1).
Ancak CHRPE olmaması GS’yi dışlamaz 2).
Osteom
Sıklık: FAP hastalarının %60-80’i
Sık görülen yerler: Çene kemiği, sinüsler, kafatası
Tanısal önemi: Üç veya daha fazlası GS’yi güçlü bir şekilde düşündürür. FAP tanısından 17 yıl önce ortaya çıkabilir
CHRPE
Sıklık: FAP hastalarının %74’ü
Özellik: Bilateral, çoklu, bezelye şeklinde. Doğumdan itibaren var olan en erken belirti
Not: CHRPE olmasa bile GS dışlanamaz
Epidermal kist
Sıklık: Yaklaşık %70
Sık görülen bölge: Baş ve boyun
Özellik: Genç yaşta ortaya çıkar ve genellikle birden fazla sayıda görülür
Desmoid tümör
Sıklık: FAP hastalarının %12-15’i
Sık görülen bölge: Karın (%37-50), ardından omuz kuşağı ve göğüs duvarı
Dikkat: FAP hastalarında ölüm nedenleri arasında kolorektal kanserden sonra ikinci sıradadır. Mortalite %10-50
FAP hastalarının %30-75’inde görülür1). Gömülü dişler ve süpernümerer dişler (FAP hastalarının %11-27’sinde, genel popülasyonda %0-4) bulunur1).
Kolorektal kanser dışında çeşitli kötü huylu tümör riskleri de artar.
| Karsinom | Yaşam boyu risk |
|---|---|
| Tiroid kanseri | FAP %2-12, Gardner tipi %10 |
| İnce bağırsak kanseri | %4-12 |
| Pankreas kanseri | Yaklaşık %1 |
(Kaynak: Alıntı 1)1)
GS’nin nedeni, APC genindeki (5q21) germ hattı mutasyonlarıdır. Mutasyonların çoğu çerçeve kayması veya anlamsız mutasyonlardır ve kesik (işlev kaybı) APC proteini üretilir.
Mutasyon bölgesi ve fenotip arasında net bir korelasyon vardır1).
| Mutasyon bölgesi | İlişkili fenotip |
|---|---|
| Kodon 311–1444 | CHRPE |
| Kodon 767-1578 | Osteom |
| Ekson 1399 ve sonrası | Desmoid (800 kat risk) |
| Kodon 1309 | 20 yaşında erken kolon kanseri |
Mutasyon küme bölgesi (MCR) kodon 1250-1464 arasındadır 1); en sık mutasyon kodon 1309’da (toplamın yaklaşık %10’u), ardından kodon 1061’de (yaklaşık %5) görülür 1). Kolorektal kanserin başlangıç yaşı da mutasyon bölgesine göre değişir: kodon 1309’da yaklaşık 20 yaş, kodon 168-1580’de yaklaşık 30 yaş, diğerlerinde ise yaklaşık 52 yaş olarak kabul edilir 1).
APC geninin moleküler genetik testi çocukluk döneminden itibaren yapılabilir. Kolonoskopi genellikle 10 yaş civarında başlatılması önerilir. Ailede APC mutasyonu biliniyorsa, mutasyonun varlığının genetik testle doğrulanması önemlidir.
Kesin tanı için APC geninin moleküler genetik testi yapılır. Yeni nesil dizileme (NGS) panel testi önerilir 1).
APC geninin moleküler genetik testi GS’nin kesin tanısı için temel oluşturur, ancak bazı hastalarda mutasyon saptanmayabilir. NGS panel testlerinin kullanımı saptama oranını artırmıştır1). Genetik test sonuçları klinik bulgularla birlikte kapsamlı olarak değerlendirilmelidir.
CHRPE’nin kendisi kötü huylu hale gelmez ve görme işlevini etkilemez. Gözle ilgili müdahale gerekmez, sadece düzenli takip yapılır.
Polip büyümesini baskılamak amacıyla ameliyat öncesinde veya hafif vakalarda kullanılır.
Tek başına ilaç tedavisi kolorektal kanser riskini ortadan kaldıramaz.
Kolektominin 25 yaşına kadar yapılması önerilir, ideal olarak 16-20 yaş arasında uygulanması tercih edilir2). Başlıca üç cerrahi yöntem vardır2).
Total Proktokolektomi + Kalıcı İleostomi
Özellik: En kesin cerrahi yöntem. Kolon epiteli kalmaz
Avantaj: Kolorektal kanser riskini maksimum düzeyde azaltır
Dezavantaj: Kalıcı stoma gerektirir ve yaşam kalitesini etkiler
Restoratif Proktokolektomi + İleal Poş-Anal Anastomoz
Özellik: Bağırsak sürekliliğini koruyan cerrahi yöntem
Avantaj: Kalıcı stomadan kaçınılabilir
Dezavantaj: Üriner ve cinsel fonksiyon komplikasyonları riski vardır
Total Kolektomi + İleorektal Anastomoz
Özellikler: Az invaziv, genç hastalar için uygun
Avantajlar: Cerrahi invazyon nispeten az
Dezavantajlar: Kalan rektumun sürekli takibi gerekli
Desmoid tümör histolojik olarak iyi huyludur ancak lokal invazyonu yüksektir ve sık tekrarlar. Tedavi multidisipliner bir yaklaşım gerektirir4)5).
Kolon rezeksiyonu sonrası aşağıdaki gözetim devam ettirilir2):
Polip sayısı ve displazi derecesine göre değişir. Az sayıda polip varsa NSAİİ veya COX-2 inhibitörleri ile ilaç tedavisi ile takip edilebilir. Ancak 20’den fazla veya yüksek dereceli displazi içeren polipler varsa profilaktik rezeksiyon önerilir. İdeal cerrahi zamanı 16-20 yaş olarak belirtilmiştir 2).
APC proteini, 2800 amino asitten oluşan bir tümör baskılayıcı faktördür. Başlıca domain yapıları olarak oligomerizasyon bölgesi, armadillo tekrarları, β-katenin bağlanma bölgesi, aksin bağlanma bölgesi ve mikrotübül bağlanma bölgesi içerir 1).
APC mutasyonu, Wnt sinyal yolunun sürekli aktivasyonuna neden olur. Normal APC proteini, β-katenini fosforile ederek proteazomda yıkımını hızlandırır. APC mutasyonunda bu işlev kaybolur, β-katenin sitoplazmada birikir ve çekirdekte TCF/LEF transkripsiyon faktörü ile kompleks oluşturarak hücre çoğalma genlerinin transkripsiyonunu artırır.
Mutasyonların çoğu çerçeve kayması veya anlamsız mutasyonlardır ve kesik (işlev kaybı) APC proteini üretilir 1).
Sporadik desmoid tümörlerde, CTNNB1 (β-katenin geni) mutasyonu %75’ten fazlasında bulunur ve benzer yol rol oynar 3).
CHRPE, retina pigment epitelinin lokal hücre çoğalma anormalliği olarak ortaya çıkar. Retina pigment epitelinin anormal çoğaldığı bölgelerde pigment yoğunlaşır ve büyür, çevredeki lakünalar (depigmente alanlar) ile kontrast oluşturarak karakteristik bir görünüm verir. Malignleşme potansiyeli neredeyse yoktur ve görme işlevi üzerinde etkisi yoktur.
Yeni nesil dizileme (NGS) panel testi, APC genindeki mutasyon bölgelerinin tanımlanma doğruluğunu artırmıştır 1). Genotip-fenotip korelasyonuna dayalı risk sınıflandırması ilerlemekte olup, mutasyon bölgesine göre kişiselleştirilmiş tarama ve tedavi stratejilerinin oluşturulması beklenmektedir 1).
Litchinko ve ark. (2022), pankreasta nadir görülen büyük bir desmoid tümör (170 mm) vakası bildirdi3). Multidisipliner ekip tarafından multidisipliner yönetim uygulandı ve pankreas desmoidinde cerrahi endikasyon ve zamanlamanın zorluğu ortaya kondu. Sorafenibin ilaç tedavisi olarak etkinliği dikkat çekmekte olup, cerrahi rezeksiyonun zor olduğu vakalarda kullanımı değerlendirilmektedir.
Yüksek yoğunluklu odaklanmış ultrason (HIFU) non-invaziv desmoid tedavisinde araştırma aşamasındadır3).
Albuquerque ve ark. (2025), omuz eklem fonksiyon rekonstrüksiyonu gerektiren bir pediatrik desmoid tümör vakası bildirdi5). Vinblastin + metotreksat ile neoadjuvan kemoterapiyi içeren multidisipliner yaklaşım, fonksiyon korunması ve tümör kontrolü sağladı. Omuz kuşağı ve göğüs duvarı desmoidlerinin tümün %37-50’sini oluşturduğu ve pediatrik vakalarda fonksiyonel rekonstrüksiyonun önemi vurgulanmaktadır.
Diaz ve ark. (2025), invaziv bazal hücreli karsinom ve total diz artroplastisi komplikasyonunu içeren cerrahi ikilem sunan bir Gardner sendromu vakası bildirerek nadir komplikasyonlarda multidisipliner yaklaşım gerekliliğini göstermiştir4). Prevalansı 100.000-160.000’de 1 olan bu hastalıkta kişiselleştirilmiş tıbbın önemi artmaktadır.