ऑस्टियोमा
आवृत्ति: FAP रोगियों में 60-80%
सामान्य स्थान: जबड़े की हड्डी, परानासल साइनस, कपाल
नैदानिक महत्व: 3 या अधिक होने पर GS का दृढ़ संकेत। FAP के निदान से 17 वर्ष पहले हो सकता है

गार्डनर सिंड्रोम (Gardner syndrome; GS) पारिवारिक एडिनोमेटस पॉलीपोसिस (familial adenomatous polyposis; FAP) का एक फेनोटाइपिक वेरिएंट है। यह बड़ी आंत के एडिनोमेटस पॉलीपोसिस के साथ-साथ ऑस्टियोमा, कोमल ऊतक ट्यूमर (डेस्मॉइड ट्यूमर), और एपिडर्मल सिस्ट जैसे एक्स्ट्राकोलोनिक अभिव्यक्तियों द्वारा विशेषता है। यह ऑटोसोमल डॉमिनेंट (पूर्व नाम: ऑटोसोमल प्रभावी) वंशानुक्रम पैटर्न का अनुसरण करता है।
कारण जीन APC (adenomatous polyposis coli) जीन है, जो गुणसूत्र 5q21 पर स्थित है। यह जर्मलाइन उत्परिवर्तन के कारण होता है, और संतानों में आनुवंशिकता की संभावना 50% है। प्रसार दर प्रति 100,000-160,000 लोगों में 1 मानी जाती है4)।
FAP के निम्नलिखित उपप्रकार हैं 1)।
FAP सभी कोलोरेक्टल कैंसर का लगभग 1% होता है1)। कोलोनिक पॉलीप्स लगभग 10 वर्ष की आयु से दिखाई देने लगते हैं और सैकड़ों से हजारों तक पहुंच जाते हैं। उपचार के बिना, 40 वर्ष की आयु में 50% और 60 वर्ष की आयु में लगभग सभी रोगियों में कोलोरेक्टल कैंसर विकसित हो जाता है। लगभग 20-30% मामलों में डी नोवो उत्परिवर्तन पाए जाते हैं1)5)।
APC जीन में लगभग 20-30% आवृत्ति पर डी नोवो उत्परिवर्तन होने के कारण, पारिवारिक इतिहास के बिना भी यह रोग विकसित हो सकता है1)5)। यह ध्यान रखना महत्वपूर्ण है कि पारिवारिक इतिहास के अभाव में GS को खारिज नहीं किया जा सकता।
बड़ी आंत के पॉलिप आमतौर पर लक्षणहीन होते हैं, और अक्सर देर से पता चलते हैं। जब लक्षण प्रकट होते हैं, तो वे मुख्य रूप से निम्नलिखित होते हैं।
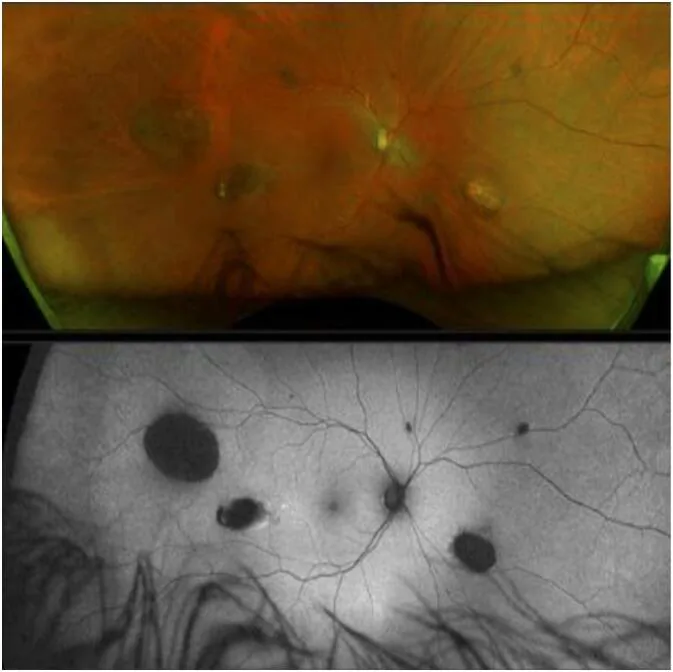
CHRPE (congenital hypertrophy of the retinal pigment epithelium) जीएस का सबसे प्रारंभिक एक्स्ट्राकोलोनिक लक्षण है, जो जन्म से मौजूद होता है 1)।
हालांकि, CHRPE की अनुपस्थिति में भी GS को खारिज नहीं किया जा सकता2)।
ऑस्टियोमा
आवृत्ति: FAP रोगियों में 60-80%
सामान्य स्थान: जबड़े की हड्डी, परानासल साइनस, कपाल
नैदानिक महत्व: 3 या अधिक होने पर GS का दृढ़ संकेत। FAP के निदान से 17 वर्ष पहले हो सकता है
CHRPE
आवृत्ति: FAP रोगियों में 74%
विशेषताएं: द्विपक्षीय, बहुवचन, मटर के आकार का। जन्म से मौजूद सबसे प्रारंभिक लक्षण
नोट: CHRPE के बिना भी GS को खारिज नहीं किया जा सकता
एपिडर्मल सिस्ट
आवृत्ति: लगभग 70%
सामान्य स्थान: सिर और गर्दन
विशेषताएँ: युवावस्था से प्रकट होता है और अक्सर कई गिनती में पाया जाता है
डेस्मॉइड ट्यूमर
आवृत्ति: FAP रोगियों का 12-15%
प्रमुख स्थान: उदर (37-50%), उसके बाद कंधे की कमर और छाती की दीवार
चेतावनी: FAP रोगियों में मृत्यु का दूसरा सबसे आम कारण कोलोरेक्टल कैंसर के बाद। मृत्यु दर 10-50%
FAP के 30-75% रोगियों में पाई जाती हैं 1)। प्रभावित दांत और अतिरिक्त दांत (FAP रोगियों में 11-27% बनाम सामान्य जनसंख्या में 0-4%) देखे जाते हैं 1)।
बड़ी आंत के अलावा अन्य विभिन्न घातक ट्यूमर का जोखिम भी बढ़ जाता है।
| कैंसर का प्रकार | आजीवन जोखिम |
|---|---|
| थायरॉइड कैंसर | FAP 2–12%, गार्डनर प्रकार 10% |
| छोटी आंत का कैंसर | 4–12% |
| अग्नाशय का कैंसर | लगभग 1% |
(स्रोत: उद्धरण 1) 1)
GS का कारण APC जीन (5q21) में जर्मलाइन उत्परिवर्तन है। अधिकांश उत्परिवर्तन फ्रेमशिफ्ट या नॉनसेंस उत्परिवर्तन होते हैं, जिसके परिणामस्वरूप छोटा (कार्य-हानि) APC प्रोटीन बनता है।
उत्परिवर्तन स्थल और फेनोटाइप के बीच स्पष्ट संबंध है 1)।
| उत्परिवर्तन स्थल | संबंधित फेनोटाइप |
|---|---|
| कोडन 311–1444 | सीएचआरपीई |
| कोडन 767–1578 | अस्थ्यर्बुद |
| एक्सॉन 1399 और उसके बाद | डेस्मॉइड (800 गुना जोखिम) |
| कोडन 1309 | 20 वर्ष की आयु में प्रारंभिक कोलोरेक्टल कैंसर |
म्यूटेशन क्लस्टर क्षेत्र (MCR) कोडन 1250 से 1464 तक है 1), सबसे आम उत्परिवर्तन कोडन 1309 (कुल का लगभग 10%) है, उसके बाद कोडन 1061 (लगभग 5%) है 1)। कोलोरेक्टल कैंसर की शुरुआत की उम्र भी उत्परिवर्तन स्थल के अनुसार भिन्न होती है: कोडन 1309 के लिए 20 वर्ष, कोडन 168-1580 के लिए लगभग 30 वर्ष, और अन्य के लिए लगभग 52 वर्ष 1)।
APC जीन का आणविक आनुवंशिक परीक्षण बचपन से ही किया जा सकता है। कोलोनोस्कोपी अक्सर 10 वर्ष की आयु के आसपास शुरू करने की सिफारिश की जाती है। यदि परिवार में APC उत्परिवर्तन ज्ञात है, तो आनुवंशिक परीक्षण द्वारा उत्परिवर्तन की उपस्थिति की पुष्टि करना महत्वपूर्ण है।
निश्चित निदान के लिए APC जीन का आणविक आनुवंशिक परीक्षण किया जाता है। अगली पीढ़ी अनुक्रमण (NGS) पैनल परीक्षण की सिफारिश की जाती है 1)।
APC遺伝子の分子遺伝学的検査はGSの確定診断の根拠となるが、一部の患者では変異が検出されない場合もある。NGSパネル検査の活用により検出率は向上している1)。遺伝子検査の結果は臨床所見と組み合わせて総合的に判断する。
CHRPEそのものは悪性化せず、視機能への影響もない。眼科的介入は不要であり、定期観察のみ行う。
ポリープの増殖抑制を目的に、手術前や軽症例に使用される。
薬物療法単独で大腸癌のリスクを排除することはできない。
大腸切除は25歳までに行うことが推奨され、理想的には16〜20歳での施行が望ましい2)。主な術式は以下の3種類である2)。
直腸結腸全摘+永久回腸瘻
特徴:最も確実な術式。大腸上皮が残存しない
利点:大腸癌リスクを最大限に低減できる
欠点:永久ストーマが必要となり生活の質に影響する
回復的直腸結腸全摘+回腸嚢肛門吻合
特徴:腸管連続性を温存する術式
利点:永久ストーマを回避できる
欠点:泌尿器・性機能合併症のリスクがある
全大腸切除+回腸直腸吻合
特徴:侵襲が少なく、若年患者向き
利点:手術侵襲が比較的少ない
欠点:残存直腸のサーベイランスが継続的に必要
デスモイド腫瘍は組織学的に良性であるが、局所浸潤性が高く再発しやすい。治療は多職種による集学的アプローチを要する4)5)。
大腸切除後も以下のサーベイランスを継続する2)。
ポリープの数や異形成の程度によって異なる。少数のポリープであればNSAIDsやCOX-2阻害薬による薬物療法で経過をみることがある。しかし、20個以上や高度異形成ポリープが認められる場合は予防的切除が推奨される。理想的な手術時期は16〜20歳とされている2)。
APCタンパク質は2800アミノ酸からなる癌抑制因子である。主要なドメイン構造として、オリゴマー化領域・アルマジロリピート・β-カテニン結合領域・アキシン結合領域・微小管結合領域を持つ1)。
APC変異はWntシグナル伝達経路の恒常的活性化を引き起こす。正常なAPCタンパク質はβ-カテニンをリン酸化してプロテアソームでの分解を促進する。APC変異ではこの機能が失われ、β-カテニンが細胞質に蓄積し、核内でTCF/LEF転写因子と複合体を形成して細胞増殖遺伝子の転写を促進する。
変異の大半はフレームシフトまたはナンセンス変異であり、切断型(機能喪失型)APCタンパク質が産生される1)。
散発性デスモイド腫瘍では、CTNNB1(β-カテニン遺伝子)変異が75%以上に認められ、同様の経路が関与する3)。
CHRPEは網膜色素上皮の局所的な細胞増殖異常として生じる。網膜色素上皮が異常増殖した部分は色素が濃縮・肥大し、周囲のラクナ(脱色素化領域)とのコントラストで特徴的な外観を呈する。悪性化ポテンシャルはほぼなく、視機能への影響も認められない。
次世代シークエンシング(NGS)パネル検査により、APC遺伝子の変異部位の同定精度が向上している1)。遺伝子型と表現型の相関に基づくリスク層別化が進み、変異部位に応じた個別化サーベイランス・治療戦略の構築が期待されている1)。
Litchinkoら(2022)は、膵臓に発生した大型デスモイド腫瘍(170 mm)の稀な1例を報告した3)。多学科チームによる集学的管理が行われ、膵臓デスモイドにおける手術適応・タイミングの難しさを提示した。薬物療法としてソラフェニブの有効性が注目されており、外科切除が困難な症例への適用が検討されている。
高密度焦点式超音波(HIFU)の非侵襲的デスモイド治療への応用も研究段階にある3)。
Albuquerqueら(2025)は、肩関節機能再建を要した小児デスモイド腫瘍の1例を報告した5)。ビンブラスチン+メトトレキサートによる術前化学療法を含む集学的アプローチにより、機能温存と腫瘍制御を達成した。肩甲帯・胸壁デスモイドが全体の37〜50%を占めるとされ、小児例における機能再建の重要性を示している。
Diazら(2025)は、浸潤性基底細胞癌と全膝関節置換術合併例を含む外科的ジレンマを呈するGS症例を報告し、稀な合併症への集学的対応が求められることを示した4)。有病率が10万〜16万人に1人とされる本疾患において、個別化医療の重要性が増している。