
網膜有髄神経線維
1. 網膜有髄神経線維とは
Section titled “1. 網膜有髄神経線維とは”網膜有髄神経線維は、通常は無髄である網膜内の視神経線維に髄鞘が限局性に形成される先天異常である。1855年にvon Jagerにより初めて報告された。有病率は約0.3〜1%とされる。
正常の有髄化と本疾患の発生
Section titled “正常の有髄化と本疾患の発生”正常では、視神経線維は視神経篩状板より後方においてのみ希突起膠細胞(オリゴデンドロサイト)による髄鞘形成を受ける。篩状板より前方、すなわち視神経乳頭から網膜内に至る部分の神経線維は無髄のままである。
正常の有髄化は胎生5か月頃に外側膝状体から視神経の方向へ向かって始まり、乳頭篩状板に到達して停止する。篩状板が希突起膠細胞の網膜内侵入を阻止する物理的バリアとして機能している。
本疾患は、希突起膠細胞が篩状板を越えて網膜内に侵入し、限局的に神経線維を有髄化することで生じる。侵入の原因については篩状板の構造異常説や発生時期のずれ説が提唱されているが、詳細は不明である。
形態的に3つの型に分類される。
| 分類 | 頻度 | 眼底所見 | 症状 |
|---|---|---|---|
| 限局性(乳頭連続型) | 最多 | 乳頭から扇形に広がる白色混濁 | 通常無症状 |
| 孤立性 | 稀 | 乳頭から離れた位置に白色混濁 | 通常無症状 |
| 広範囲型 | まれ | 広範な白色混濁 | 視力障害・視野欠損 |
2. 主な症状と臨床所見
Section titled “2. 主な症状と臨床所見”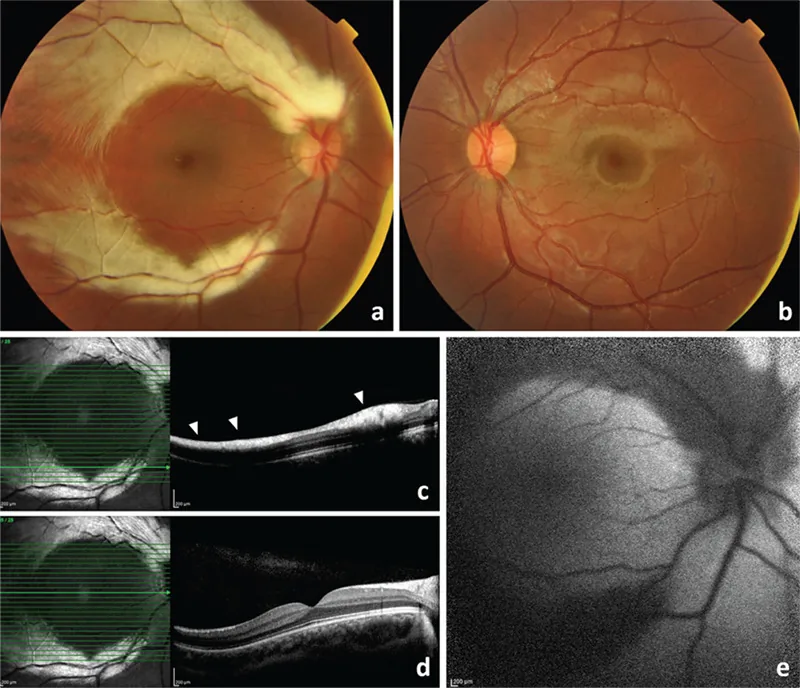
大多数の症例は限局性で小領域のため無症状である。眼底検査で偶然指摘されることが多い。広範囲型では視力障害や視野欠損をきたす。
特徴的な所見は網膜神経線維の走行に沿った刷毛状(feathered edge)の白色混濁である。辺縁は羽毛状に不規則で、神経線維束の走行と一致する。
- 乳頭連続型(最多): 視神経乳頭から扇形に広がる白色混濁。乳頭を中心に放射状に走行する神経線維に沿う
- 孤立型: 乳頭から離れた位置に孤立して存在。周囲との境界は羽毛状
- 広範囲型: 広範な白色混濁で視力や視野に影響する
各種撮影所見
Section titled “各種撮影所見”- 赤外光眼底撮影・レッドフリー眼底撮影: 髄鞘中に脂質が多く含まれるため白色として認識される
- 自発蛍光眼底撮影(FAF): 正常な自発蛍光が遮断されるため低蛍光を示す
- 蛍光眼底造影(FA): 背景蛍光をブロックし、病巣への蛍光漏出もみられない(軟性白斑と異なり蛍光漏出なし)
- OCT: 網膜神経線維層(RNFL)の厚みが有髄化部位に一致して異常高値を示す
広範囲型では以下の合併所見を認めることがある。
Straatsma症候群
Section titled “Straatsma症候群”有髄神経線維+高度近視+弱視の三徴を呈する症候群をStraatsma症候群(または広範囲有髄神経線維症候群)と呼ぶ。広範囲型で認められる概念であり、視機能への影響が大きい。まれな病態である。
広範囲型の有髄神経線維を有する小児では、弱視の早期発見と対処が重要課題となる。ただし弱視治療の効果は乏しく、最終的な視力予後は不良なことが多い。高度近視については屈折矯正で対応するが、軸性近視の進行を伴う場合は近視性合併症のリスクにも注意して経過観察する。
3. 原因とリスク要因
Section titled “3. 原因とリスク要因”先天性異常としての位置づけ
Section titled “先天性異常としての位置づけ”網膜有髄神経線維は先天性の発生異常であり、後天的な原因により発症するものではない。特定のリスク因子は知られていない。遺伝性については家族性の報告が散見されるものの稀であり、遺伝形式は確立していない。
まれに遺伝性症候群(GAPO症候群、オールブライト遺伝性骨ジストロフィー、神経線維腫症1型など)との関連が報告されているが、大多数は症候群を伴わない孤発例である。後天性の有髄神経線維は極めてまれであり、鈍的外傷、視神経乳頭ドルーゼン、視神経炎後などとの関連が報告されている。
胎生5か月頃に外側膝状体から始まる視神経の有髄化が、篩状板で停止せず網膜内に進展することで生じる。篩状板の物理的バリア機能が何らかの理由で障害された場合、あるいは有髄化の開始時期が早い場合に生じると推測されている。
有髄化が視機能に与える影響は、髄鞘の白色不透明性による光の透過障害である。髄鞘は光の透過を妨げ、相対暗点を形成する。広範囲型では視力低下や視野欠損の原因となる。弱視の機序は、髄鞘による光遮断が発達期の視覚入力を制限することによる形態覚遮断弱視に類似した状態と考えられる。
4. 診断と検査方法
Section titled “4. 診断と検査方法”特徴的な眼底所見により診断する。神経線維の走行に沿った刷毛状の白色混濁が典型的であり、辺縁が羽毛状であることが鑑別の重要なポイントとなる。診断に追加の検査を要することは少ないが、広範囲型では鑑別疾患を除外するために画像検査や視野検査を実施する。
| 所見 | 網膜有髄神経線維 | 軟性白斑 |
|---|---|---|
| 反射 | 強い(刷毛状・光沢あり) | 弱い(綿花状) |
| 浮腫 | なし(平坦) | あり(隆起) |
| 網膜血管との関係 | 太い血管が覆われる | 覆われない |
| FA所見 | 背景蛍光ブロックのみ・漏出なし | 虚血性変化・漏出あり |
| 場所 | 神経線維走行に一致 | 血管走行周囲 |
その他の鑑別が必要な疾患として、小児の白色眼底病変では網膜芽細胞腫(腫瘤性・隆起性、石灰化を伴う)、アストロサイトーマ(結節性硬化症に伴う白色網膜結節)が挙げられる。
- 眼底検査(直接・倒像検眼鏡): 特徴的な白色混濁の確認
- 蛍光眼底造影: 蛍光漏出の有無を確認(軟性白斑との鑑別)
- 自発蛍光撮影: 低蛍光パターンの確認
- 赤外光撮影・レッドフリー撮影: 白色混濁のコントラスト評価
- OCT: RNFL厚の異常高値確認(有髄化部位に一致)
- 視野検査: 広範囲型で適応。対応する暗点の評価
軟性白斑(神経線維走行に沿わず蛍光漏出あり)、網膜芽細胞腫(小児、腫瘤性、石灰化あり)、アストロサイトーマ(結節性硬化症に伴う白色網膜結節)などの鑑別が必要である。有髄神経線維は反射が強く平坦で、蛍光眼底造影で蛍光漏出を伴わない点が鑑別の要点となる。
5. 標準的な治療法
Section titled “5. 標準的な治療法”原則: 治療不要
Section titled “原則: 治療不要”大多数の症例は限局性、無症状であるため治療を要しない。経過観察のみで十分であり、定期的な眼科受診によって合併症の有無を確認する。非進行性の先天異常であることを患者、保護者に十分説明し、不要な不安を与えないことも重要な診療上の対応である。
経過観察の目安
Section titled “経過観察の目安”定期的な眼底検査により病変の変化を記録する。通常は大きさや範囲の拡大を認めないが、以前の記録と比較して病変が縮小、消失している場合は視神経損傷の可能性を考慮し精査を行う。小児では弱視のリスク評価のため、視力測定と屈折検査を定期的に行う。髄鞘の消失は網膜神経損傷のサインとなりうるため、ベースライン眼底画像の保存が推奨される。
合併症への対応
Section titled “合併症への対応”- 弱視: 遮蔽療法(健眼パッチング)などの弱視治療が試みられることがある。しかし効果はほとんどないとされており、治療開始の判断には慎重な対応が必要である
- 高度近視: 適切な屈折矯正(眼鏡またはコンタクトレンズ)を行う。不同視が強い場合はコンタクトレンズが有利なことがある
- 網膜血管異常: 合併する血管異常の種類に応じた対応。新生血管や硝子体出血を合併した場合はレーザー光凝固が行われることがある
- 斜視: 合併する斜視については通常のプロトコルに従い管理する
6. 病態生理学・詳細な発症機序
Section titled “6. 病態生理学・詳細な発症機序”正常の視神経有髄化過程
Section titled “正常の視神経有髄化過程”正常の視神経有髄化は胎生5か月頃に外側膝状体から始まる。有髄化は外側膝状体、視放線、視交叉、視神経、篩状板の順に進行し、篩状板に到達したところで停止する。篩状板が物理的バリアとして希突起膠細胞の網膜内侵入を阻止するため、正常では網膜内の神経線維は無髄のまま維持される。
本疾患の発生機序
Section titled “本疾患の発生機序”希突起膠細胞が乳頭篩状板を越えて網膜内に侵入し、網膜神経線維を限局的に有髄化することで本疾患が生じる。限局的侵入の原因は不明であり、以下の説が提唱されている。
- 篩状板構造異常説: 篩状板に部分的な欠損や構造異常があり、希突起膠細胞の侵入を許す
- 発生時期のずれ説: 有髄化の開始時期が正常より早く、篩状板が形成される前に希突起膠細胞が網膜内に侵入する
組織病理学的特徴
Section titled “組織病理学的特徴”組織学的には以下の知見が得られている[6]。
- 有髄神経線維の領域内では、有髄線維と無髄線維が混在する。特定の斑状領域や神経束に限定されず、無髄線維束の間に単一の有髄線維が散在する。
- 領域内の有髄・無髄線維はいずれも、正常網膜の線維よりも直径が大きい。
- 有髄化領域では網膜神経節細胞の集団が減少しており、その下層の内網状層・外網状層の厚みも減少している。
- 細胞核は比較的少なく、顕微鏡的な炎症所見は認められない。
- 肉眼的に視神経乳頭と連続して見える病変でも、組織学的には視神経の髄鞘形成領域と連続していない場合がある。
視機能への影響
Section titled “視機能への影響”髄鞘は白色不透明であり、光の透過を妨げる。限局性の有髄神経線維は小さな相対暗点を形成するが、脳の可塑性により自覚症状に乏しいことが多い。広範囲型では視力低下、視野欠損の原因となる。弱視は形態覚遮断弱視に類似した機序で生じると考えられており、発達期に有髄化が光の透過を妨げることで視覚入力が制限される。弱視治療の効果が乏しいことは、この形態覚遮断の程度が大きいことを示唆している。
OCTにおける注意点
Section titled “OCTにおける注意点”OCTによる評価では、有髄化部位のRNFL厚が異常に増加しているため、緑内障の診断においてRNFL厚の過大評価(セグメンテーションエラー)を引き起こしうる。有髄神経線維を有する眼では緑内障による真のRNFL菲薄化が隠される可能性があり、OCT結果の解釈に特に注意が必要である。視野検査や眼圧測定などの他の検査所見を合わせて総合的に判断する。
緑内障眼では一般にOCTの自動セグメンテーションで補正を要する頻度が高いことが報告されており、有髄神経線維のように反射が強く肥厚した領域では自動解析のみに依存することのリスクが指摘されている[8]。読影時にはBスキャン断層像を確認し、必要に応じて手動補正を行う。
有髄神経線維の消失とその原因
Section titled “有髄神経線維の消失とその原因”有髄神経線維は通常は静止性であるが、以下の疾患で消失が報告されている。消失は網膜神経軸索の病理学的変性を反映すると考えられている[6]。
- 神経疾患: 下垂体腺腫、視神経炎、急性視神経症、原発開放隅角緑内障(POAG)[7]
- 炎症性疾患: ベーチェット病による乳頭炎・硝子体炎
- 網膜疾患: 網膜分枝動脈閉塞症(BRAO)、網膜中心動脈閉塞症(CRAO)、糖尿病網膜症
- 医原性: 脈絡膜メラノーマに対する小線源治療、網膜前膜に対する硝子体手術
以前の記録で確認されていた有髄神経線維が消失している場合は、これらの背景疾患による網膜神経損傷を疑い精査する。
本疾患は非進行性の先天異常であり、通常は経時的に有髄化の範囲が拡大することはない。限局性、孤立性の症例では視機能への影響は乏しく、長期的な予後は良好である。広範囲型では視力低下、視野欠損、弱視が持続し、弱視治療の効果は乏しいため視機能への影響が残存する。高度近視を合併する場合は長期フォローが必要となる。
7. 最新の研究と今後の展望
Section titled “7. 最新の研究と今後の展望”OCTによる網膜有髄神経線維の定量評価研究が進んでいる。RNFL厚の異常高値パターンが報告されており(有髄化部位の平均RNFL厚は健眼の約114μmに対し152μm前後と有意に肥厚するとの小児コホート報告がある)、診断補助としての有用性が検討されている。また緑内障診断におけるOCT評価でRNFL厚が過大評価される可能性があり、有髄神経線維を有する眼での読影には注意が必要とされている。
多発性硬化症や視神経炎後に有髄神経線維が退縮した症例報告がある。先天性の有髄神経線維が炎症や脱髄疾患によって消失しうることは、本疾患の自然経過に関する理解を深めるうえで重要な知見である。
Straatsma症候群の疫学、機序については定量的研究が少なく、さらなる多施設データの集積が求められている。長期追跡研究では弱視治療への反応性が乏しい一方で、不同視矯正と積極的遮蔽療法の併用により部分的な視力改善を得た例も報告されており、早期介入の意義については議論が続いている。遺伝学的背景についても現時点では不明な点が多い。
全ゲノムシーケンス
Section titled “全ゲノムシーケンス”網膜有髄神経線維の患者に対して全ゲノムシーケンスが実施され、疾患関連遺伝子変異や意義不明の新規変異が同定されている。本疾患は多くの遺伝子変異の累積的効果から生じる可能性が示唆されている。この研究では、当該患者が視神経乳頭異常および網膜剥離のリスクが高い一方、加齢黄斑変性に対する遺伝的素因は平均以下であることも示された[6]。
新しいイメージング技術(補償光学)
Section titled “新しいイメージング技術(補償光学)”補償光学(adaptive optics)を用いた経強膜光位相イメージング法が開発されている。この手法により、非侵襲的に2〜3マイクロメートルの解像度で有髄神経線維を観察できるようになった[6]。
先天異常であり、通常は非進行性である。大きさや範囲が拡大することは基本的にない。ただし、多発性硬化症や視神経炎の後に有髄神経線維が消失(退縮)した症例報告がある。先天性有髄神経線維が真の意味で安定かについての再検討も行われている。
-
Tarabishy AB, Alexandrou TJ, Traboulsi EI. Syndrome of myelinated retinal nerve fibers, myopia, and amblyopia: a review. Surv Ophthalmol. 2007 Nov-Dec;52(6):588-96. PMID: 18029268
-
Kee C, Hwang JM. Visual prognosis of amblyopia associated with myelinated retinal nerve fibers. Am J Ophthalmol. 2005 Feb;139(2):259-65. PMID: 15733986
-
Shen Y, Zhao J, Sun L, Zeng L, Chen Z, Tian M, Zhou X. The long-term observation in Chinese children with monocular myelinated retinal nerve fibers, myopia and amblyopia. Transl Pediatr. 2021 Apr;10(4):860-869. PMID: 34012835
-
Kera J, Fasiuddin AF. Ocular Findings Associated With Myelinated Retinal Nerve Fibers. Cureus. 2021 Apr 19;13(4):e14552. PMID: 34017666 / PMCID: PMC8130640
-
Sevik MO, Aykut A, Karaman NF, Şahin Ö. Straatsma Syndrome: Should Visual Prognostic Factors Be Taken into Account? A Case Report. Turk J Ophthalmol. 2021 Dec 28;51(6):398-402. PMID: 34963270 / PMCID: PMC8715659
-
Ramkumar HL, Verma R, Ferreyra HA, Robbins SL. Myelinated Retinal Nerve Fiber Layer (RNFL): A Comprehensive Review. Int Ophthalmol Clin. 2018;58(4):147-156. PMID: 30239369
-
Sowka JW, Nadeau MJ. Regression of myelinated retinal nerve fibers in a glaucomatous eye. Optom Vis Sci. 2013 Jul;90(7):e218-e220. PMID: 23708924
-
Mansberger SL, Menda SA, Fortune BA, Gardiner SK, Demirel S. Automated Segmentation Errors When Using Optical Coherence Tomography to Measure Retinal Nerve Fiber Layer Thickness in Glaucoma. Am J Ophthalmol. 2017;174:1-8. PMID: 27818206