L’épilepsie myoclonique avec fibres rouges déchiquetées (MERRF) est une maladie mitochondriale multisystémique rare due à une mutation de l’ADN mitochondrial. Dans la CIM-10, elle est classée sous E88.4 (troubles du métabolisme mitochondrial).
Environ 80 % des cas de MERRF sont causés par une mutation ponctuelle m.8344A>G du gène MT-TK5). Cette mutation entraîne un dysfonctionnement de l’ARNt lysine, perturbant l’assemblage des protéines nécessaires à la phosphorylation oxydative1). D’autres mutations causales rapportées incluent m.8356T>C, m.8363G>A et m.3243A>G5). Une nouvelle mutation du gène MT-TC (m.5820C>A) a également été rapportée2).
Dans les cas héréditaires, la transmission est presque exclusivement maternelle. Les trois principales maladies représentatives des encéphalomyopathies mitochondriales sont l’ophtalmoplégie externe progressive chronique (CPEO), le MELAS et le MERRF, le MELAS étant le plus fréquent.
La prévalence du MERRF est estimée à moins d’un cas pour 100 000 personnes. L’âge moyen d’apparition est de 45 ans5), mais certains cas se manifestent dès la petite enfance par des crises d’épilepsie. Il a été rapporté que 92,3 % des porteurs de la mutation m.8344A>G développent une épilepsie4).
Le degré d’hétéroplasmie (proportion d’ADNmt mutant et sauvage dans une même cellule) influence considérablement l’expression clinique1)3). Lorsque la charge mutationnelle est faible, des formes bénignes sans symptômes du système nerveux central sont possibles. Des familles n’ayant présenté aucun symptôme clinique du système nerveux central sur quatre générations ont également été rapportées3).
QComment le MERRF est-il hérité ?
A
L’ADN mitochondrial étant hérité uniquement de la mère, il suit un mode de transmission maternelle. La proportion d’ADNmt muté (hétéroplasmie) varie selon les individus et les tissus, influençant la sévérité des symptômes cliniques. Voir la section « Causes et facteurs de risque » pour plus de détails.
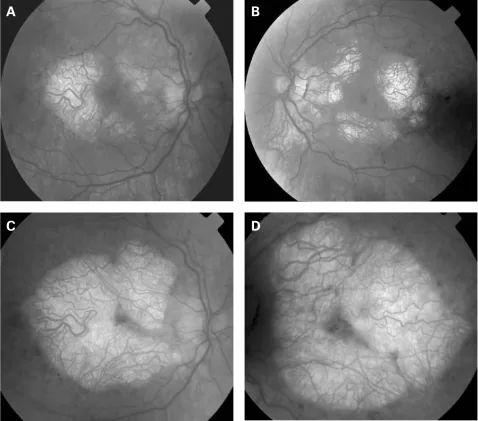
P P Rath, S Jenkins, M Michaelides et al. Characterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence. The British Journal of Ophthalmology. 2008 Jan 22; 92(5):623. Figure 2. PMCID: PMC2569141. License: CC BY.
Figure 1 : Évolution de l’acuité visuelle chez des patients atteints du syndrome MERRF entre 1994 et 2001
Les quatre signes classiques du MERRF sont les myoclonies, l’épilepsie généralisée, l’ataxie et les fibres rouges déchiquetées à la biopsie musculaire. En neuro-ophtalmologie, les symptômes suivants sont les principales plaintes.
Baisse de l’acuité visuelle : survient progressivement avec l’atrophie optique. Elle est souvent bilatérale et symétrique.
Myoclonies : contractions et relâchements musculaires rapides involontaires, durant généralement 1 à 2 secondes5). Elles peuvent être déclenchées par la stimulation lumineuse2).
Crises épileptiques : les crises myocloniques sont les plus fréquentes, mais des crises tonico-cloniques généralisées, des crises focales et des absences ont également été rapportées5).
Ataxie cérébelleuse : ataxie et dysarthrie apparaissent de façon progressive4).
Surdité de perception : complication fréquente.
Engourdissements périphériques : troubles sensitifs distaux prédominants liés à une neuropathie périphérique2).
Faiblesse musculaire et fatigabilité : dues à une myopathie4).
Les principaux signes neuro-ophtalmologiques associés au MERRF sont l’atrophie/neuropathie optique, l’ophtalmoplégie, le ptosis, la rétinopathie pigmentaire et le nystagmus.
Atrophie optique
Fréquence : 39 % des patients atteints de MERRF (Hirano et al., 13/36 cas), 10 % de l’ensemble des porteurs de la mutation m.8344A>G (Altmann et al., 34 cas)1)
Signes : Pâleur bilatérale et symétrique de la papille optique1). Une étude rapporte une atrophie optique chez 6 des 7 enfants et jeunes adultes confirmés génétiquement (85,7 %).
Point d’attention : Un amincissement de la RNFL et du GCC maculaire a été détecté chez les 3 patients asymptomatiques sur le plan visuel.
Ophtalmoplégie et ptosis
Ophtalmoplégie : 2 cas sur 34 (5,9 %). Les rapports sur la limitation des mouvements oculaires externes varient : 2/2 cas (Zhu et al.) contre 3/7 cas (42,9 %, Grönlund et al.).
Ptosis : 10 cas sur 34 (29,4 %, Mancuso et al.). 1 cas sur 7 (Grönlund et al.).
Rétinopathie et nystagmus
Rétinopathie pigmentaire : 4 cas sur 24 (16,7 %, Mancuso et al.). Dystrophie rétinienne chez 1 cas sur 7 (Grönlund et al.).
QY a-t-il des anomalies du nerf optique même si l'acuité visuelle est normale ?
A
Même chez les patients asymptomatiques sur le plan visuel, l’OCT peut détecter un amincissement de la couche des fibres nerveuses rétiniennes et du complexe de cellules ganglionnaires maculaires. Cela reflète une perte potentielle de cellules nerveuses rétiniennes, et un suivi ophtalmologique régulier est recommandé.
Fibres rouges déchiquetées : retrouvées dans 96 % des cas de mutation m.8344A>G par biopsie musculaire1).
Lipomatose multiple : prévalence variant de 3 à 32,4 % selon les études1). Une association entre un haut niveau d’hétéroplasmie et la taille des lipomes a été suggérée1).
Cardiomyopathie, diabète, hypothyroïdie : complications dans le cadre d’une atteinte multi-organique5).
Acidose lactique : augmentation du lactate dans le liquide céphalorachidien2).
Déficience intellectuelle / démence : apparaît dans les cas évolués.
La cause du MERRF est une mutation de l’ADN mitochondrial (ADNmt).
Mutation m.8344A>G du gène MT-TK : la plus fréquente, représentant environ 80 % des cas5). Elle provoque une anomalie structurale de l’ARNt lysine, altérant la synthèse des protéines nécessaires à la phosphorylation oxydative1).
Autres mutations de l’ADNmt : m.8356T>C, m.8363G>A, m.3243A>G, m.3255G>A, m.5820C>A du gène MT-TC, etc.2)5).
Hérédité maternelle : l’ADNmt étant transmis uniquement par la mère, la maladie ne se propage que dans la lignée maternelle des familles atteintes.
Hétéroplasmie et effet de seuil : lorsque la proportion d’ADNmt mutant dépasse un seuil spécifique au tissu, des anomalies biochimiques apparaissent3). Le seuil dans le muscle squelettique est estimé entre 60 et 90 % de mutation ; si environ 15 % d’ADNmt sauvage subsiste, la traduction et l’activité COX sont presque normales3).
Gènes modificateurs nucléaires : des facteurs influençant la ségrégation et la réplication de l’ADNmt, régulant la distribution de la charge mutationnelle entre les tissus, ont été suggérés3).
Les tests de génétique moléculaire sont essentiels pour un diagnostic définitif. La mutation m.8344A>G est identifiée chez plus de 80 % des patients5).
Échantillon : extraction d’ADN à partir du sang, de l’urine ou du muscle
Méthode : quantification de l’hétéroplasmie par PCR-RFLP3)
Remarque : l’hétéroplasmie urinaire est généralement plus élevée que celle du sang3)
La coloration trichrome de Gomori modifiée permet de visualiser des fibres rouges déchiquetées. On observe une forte coloration SDH et des fibres déficientes en COX. La double coloration COX-SDH montre environ 5 % de fibres déficientes en COX 2). La microscopie électronique révèle une prolifération et un allongement anormaux des mitochondries sous le sarcolemme 2).
Tomographie par cohérence optique (OCT) : un amincissement de la couche des fibres nerveuses rétiniennes et du complexe des cellules ganglionnaires rétiniennes peut être détecté même chez les patients asymptomatiques1)
Examen du fond d’œil : pâleur de la papille optique1)
Au début, elle est souvent confondue avec le JME, et les symptômes systémiques associés tels que l’ataxie et la progression du déclin cognitif sont utiles pour le diagnostic différentiel de la PME1).
Il n’existe actuellement aucun traitement curatif pour le MERRF. Le traitement est principalement symptomatique, et une approche multidisciplinaire combinant la gestion de l’épilepsie et les soins de soutien systémiques est la base.
Le lévétiracétam est considéré comme le plus efficace contre l’épilepsie myoclonique 5).
Traitement combiné lévétiracétam + clonazépam : Dans une étude portant sur 17 patients atteints de MERRF, une amélioration a été observée chez les 12 patients ayant changé de monothérapie à la bithérapie. Des améliorations des fonctions cognitives et de la coordination motrice ont également été rapportées 5).
Clonazépam : En tant que benzodiazépine, il est efficace seul contre les myoclonies 5).
Zonisamide : Son efficacité a été suggérée 5).
Piracétam : Efficace dans certains cas 5). Chez une femme japonaise de 68 ans, après l’arrêt de la phénytoïne et de la carbamazépine, le passage à un traitement principalement à base de piracétam et de lévétiracétam a amélioré les symptômes 2).
ASM
Effet sur les myoclonies
Précautions
Lévétiracétam
Efficace
Première intention
Clonazépam
Efficace
Sédation
Zonisamide
Potentiellement efficace
—
Piracétam
Potentiellement efficace
—
Acide valproïque
—
Contre-indication de principe (toxicité mitochondriale)
Il est recommandé de maintenir la capacité motrice par la physiothérapie et les exercices aérobiques. L’ergothérapie et l’orthophonie sont également utiles4).
Un suivi régulier en neurologie, ophtalmologie, cardiologie et endocrinologie est nécessaire1). Une évaluation clinique, des analyses sanguines, un EEG et une surveillance des taux sanguins d’ASM tous les trois mois sont recommandés5).
QPourquoi le valproate ne peut-il pas être utilisé dans le MERRF ?
A
Il a été expérimentalement confirmé que le valproate réduit l’activité des complexes I et IV de la chaîne respiratoire mitochondriale, ce qui présente un risque d’aggravation de la pathologie dans les maladies mitochondriales comme le MERRF5). Pour le traitement de l’épilepsie, on privilégie des médicaments à faible toxicité mitochondriale, tels que le lévétiracétam ou le clonazépam.
6. Physiopathologie et mécanisme détaillé de la maladie
La mutation m.8344A>G du gène MT-TK modifie la structure de l’ARNt lysine, perturbant la synthèse protéique mitochondriale (assemblage des complexes de phosphorylation oxydative)1). Il en résulte une diminution de la production d’ATP, affectant préférentiellement les organes à forte demande énergétique (cerveau, muscle squelettique, myocarde)3).
Les mutations de l’ADN mitochondrial existent à l’état d’hétéroplasmie, et lorsque la proportion d’ADNmt mutant dépasse un seuil spécifique au tissu, un phénotype biochimique apparaît 3).
Seuil pathologique du muscle squelettique : 60 à 90 % de mutation 3)
Avec environ 15 % d’ADNmt sauvage résiduel, la traduction et l’activité de la COX peuvent être restaurées à des niveaux presque normaux 3)
Les gènes modificateurs nucléaires régulent la ségrégation et la réplication de l’ADNmt de manière spécifique au tissu 3)
En réaction compensatoire au déficit en COX (déficit du complexe IV de la chaîne respiratoire), des mitochondries anormales prolifèrent sous le sarcolemme, formant des fibres rouges déchiquetées (RRF) 2). La microscopie électronique révèle une prolifération marquée de mitochondries anormalement allongées et des inclusions paracristallines 2).
Les cellules ganglionnaires de la rétine ont des besoins énergétiques élevés et sont vulnérables au dysfonctionnement mitochondrial 1). Même chez les patients asymptomatiques sur le plan visuel, un amincissement du RNFL et du GCC est détecté, suggérant une perte neuronale sous-jacente progressive.
Le dysfonctionnement mitochondrial entraîne une augmentation du stress oxydatif, des anomalies du système immunitaire et des modifications de la dynamique mitochondriale, et participe également à la physiopathologie des crises d’épilepsie4).
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Baysal et al. (2025) ont appliqué un protocole de titration rapide (augmentation jusqu’à 2 mA en 6 jours) d’un dispositif de stimulation du nerf vague (VNS) chez une femme de 38 ans présentant une mutation m.8344A>G et un état de mal épileptique super-réfractaire (SRSE)4). Sept jours après l’implantation du VNS, une amélioration des myoclonies a été observée, et 21 jours plus tard, les crises tonico-cloniques généralisées ont disparu. Lors d’un suivi de deux ans, aucune récidive de crises tonico-cloniques généralisées ni d’état de mal épileptique n’a été notée, et les myoclonies ont diminué. Le mécanisme d’action du VNS impliquerait l’activation de la voie noyau du tractus solitaire → locus coeruleus (libération de noradrénaline) et noyau du raphé (libération de sérotonine) via la stimulation du nerf vague gauche, ainsi qu’une régulation à la hausse de la densité des récepteurs GABA-A4).
Le taux d’arrêt de l’SE par VNS est rapporté à 74 % (28 cas sur 38) dans une revue systématique, mais un possible biais de publication a été souligné4).
Possibilités de nouveaux traitements médicamenteux
Le pérampanel et le rufinamide ont montré une efficacité dans l’épilepsie myoclonique non mitochondriale, et leur application au MERRF est attendue5).
Parmi les autres thérapies alternatives, le régime Atkins, les glucocorticoïdes, le cannabidiol (CBD), la N-acétylcystéine, la stimulation cérébrale profonde et la stimulation magnétique transcrânienne (réduction de 30 à 40 % de la fréquence des crises) sont à l’étude5).
Kawazoe et al. (2022) ont rapporté le cas d’une femme japonaise de 68 ans présentant une mutation homoplasmique m.5820C>A dans le gène MT-TC2). Cette mutation est située à la base de la tige du récepteur d’acides aminés de l’ARNt cystéine, et sa pathogénicité a été prédite par analyse in silico. Dans ce cas, une biopsie cutanée a révélé des inclusions intranucléaires positives pour p62, suggérant un nouveau lien entre les maladies mitochondriales et les inclusions intranucléaires2).
QQu'est-ce que la stimulation du nerf vague comme traitement ?
A
Il s’agit d’une méthode thérapeutique qui consiste à enrouler une électrode autour du nerf vague cervical gauche et à envoyer périodiquement des impulsions électriques à partir d’un stimulateur implanté sous la peau de la poitrine. Elle exerce un effet anticonvulsivant via le tronc cérébral. Un rapport de cas a montré son efficacité contre l’état de mal épileptique super-réfractaire dans le cadre du MERRF4), mais les preuves sont limitées.
Jeeva-Patel T, Freund P, Margolin EA. Lipomatosis and optic neuropathy clinches the diagnosis of myoclonic epilepsy with ragged red fibres (MERRF) syndrome. BMJ Case Rep. 2021;14:e240463.
Kawazoe T, Tobisawa S, Sugaya K, et al. Myoclonic epilepsy with ragged-red fibers with intranuclear inclusions. Intern Med. 2022;61:547-552.
Ripolone M, Zanotti S, Napoli L, et al. MERRF mutation A8344G in a four-generation family without central nervous system involvement: clinical and molecular characterization. J Pers Med. 2023;13:147.
Baysal L, Jobi S, Zimmermann S, Helmers A-K, Margraf NG. Successful application of vagus nerve stimulation in super refractory status epilepticus associated with MERRF syndrome. Epilepsy Behav Rep. 2025;30:100769. doi:10.1016/j.ebr.2025.100769.
Finsterer J. A review of the advances in the medical management of epilepsy associated with myoclonic epilepsy with ragged-red fibers (MERRF) syndrome. Cureus. 2025;17(4):e82875.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.