視神經萎縮
頻率:MERRF患者中39%(Hirano等人13/36例),m.8344A>G突變攜帶者整體中10%(Altmann等人34例)1)
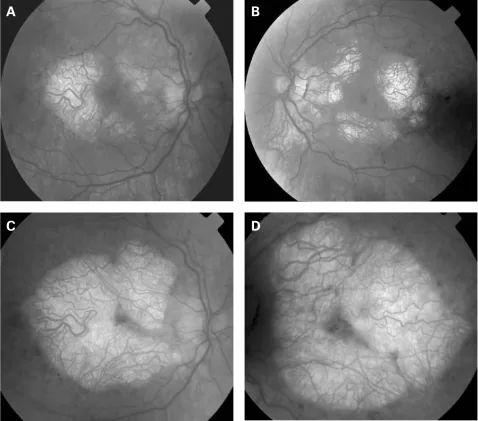
表現:雙側、對稱性視神經盤蒼白1)。有報告指出,經遺傳學確診的7例兒童及年輕成人中,6例(85.7%)有視神經萎縮。
注意點:所有3例視覺無症狀患者均檢測到RNFL及黃斑GCC變薄。

伴有破碎紅纖維的肌陣攣癲癇(MERRF)是一種由粒線體DNA突變引起的罕見多系統粒線體疾病。ICD-10分類為E88.4(粒線體代謝障礙)。
MERRF約80%由MT-TK基因的m.8344A>G點突變引起5)。此突變導致tRNA賴氨酸功能異常,損害氧化磷酸化所需蛋白質的組裝1)。其他致病突變包括m.8356T>C、m.8363G>A、m.3243A>G等5)。MT-TC基因的新突變(m.5820C>A)也有報導2)。
遺傳形式幾乎均為母系遺傳。粒線體腦肌病的代表性疾患包括慢性進行性外眼肌麻痺(CPEO)、MELAS和MERRF,其中MELAS最常見。
MERRF的盛行率估計低於每10萬人1例。平均發病年齡為45歲5),但也有在嬰兒早期以癲癇發作發病的病例。據報導,m.8344A>G突變攜帶者中92.3%會出現癲癇4)。
異質性(同一細胞內突變型與野生型mtDNA的混合比例)程度會顯著影響臨床表現1)3)。若突變量低,可能出現缺乏中樞神經系統症狀的輕症型。也有報告指出,一個家族歷經四代均未出現中樞神經系統臨床症狀3)。
粒線體DNA僅由母親遺傳,因此呈現母系遺傳模式。突變型mtDNA的比例(異質性)因人而異,也因組織而異,影響臨床症狀的嚴重程度。詳情請參閱「原因與風險因素」一節。
MERRF的典型四聯徵包括肌陣攣、全身性癲癇、共濟失調以及肌肉活檢發現的破碎紅纖維。在神經眼科領域,以下症狀為主要主訴。
MERRF相關的主要神經眼科表現包括視神經萎縮/視神經病變、眼肌麻痺、眼瞼下垂、色素性視網膜病變及眼球震顫等五項。
視神經萎縮
頻率:MERRF患者中39%(Hirano等人13/36例),m.8344A>G突變攜帶者整體中10%(Altmann等人34例)1)
表現:雙側、對稱性視神經盤蒼白1)。有報告指出,經遺傳學確診的7例兒童及年輕成人中,6例(85.7%)有視神經萎縮。
注意點:所有3例視覺無症狀患者均檢測到RNFL及黃斑GCC變薄。
眼肌麻痺・眼瞼下垂
眼肌麻痺:34例中2例(5.9%)。外眼肌運動受限的報告比例不一,Zhu等人為2/2例,Grönlund等人為7例中3例(42.9%)。
眼瞼下垂:34例中10例(29.4%,Mancuso等人)。7例中1例(Grönlund等人)。
視網膜病變・眼球震顫
色素性視網膜病變:24例中4例(16.7%,Mancuso等人)。7例中1例有視網膜失養症(Grönlund等人)。
眼震:7例中有1例(Grönlund等人)。
即使視覺無症狀的患者,OCT檢查也可能檢測到視網膜神經纖維層或黃斑部神經節細胞複合體變薄。這反映了潛在的視網膜神經細胞喪失,建議定期眼科追蹤。
MERRF的原因為粒線體DNA(mtDNA)突變。
MERRF的診斷採用以下四項臨床特徵(典型四徵)。
確定診斷需要進行分子遺傳學檢查。超過80%的患者可發現m.8344A>G突變5)。
使用Gomori三色改良染色法可確認破碎紅纖維。可見到SDH強染色、COX缺失纖維。在COX-SDH雙重染色中,約5%的纖維呈現COX缺失2)。電子顯微鏡下可觀察到肌膜下粒線體異常增生及延長2)。
乳酸19 mg/dL(正常4.2~17.0)、丙酮酸1.2 mg/dL(正常0.3~0.9)輕度升高2)。
骨骼肌的complex IV活性降至對照組的55~69%2)。
MERRF是進行性肌陣攣癲癇(PME)的一種類型,需與以下疾病進行鑑別。
| 疾病 | 鑑別要點 |
|---|---|
| 青少年肌陣攣癲癇(JME) | 無認知功能下降或共濟失調進展,預後良好 |
| Lafora病 | 皮膚活檢可見Lafora小體陽性 |
| Unverricht-Lundborg病 | CSTB基因突變、進展緩慢 |
| 神經元蠟樣脂褐質沉積症 | 視網膜變性顯著、酵素缺乏 |
初期容易誤診為JME,失調或認知功能下降等全身性伴隨症狀有助於鑑別PME1)。
目前MERRF尚無根治性治療。治療以症狀治療為主,結合癲癇管理與全身支持療法的多專科協作是基本原則。
對於肌陣攣性癲癇,左乙拉西坦被認為最有效5)。
| ASM | 對肌陣攣的效果 | 注意事項 |
|---|---|---|
| 左乙拉西坦 | 有效 | 第一線 |
| 氯硝西泮 | 有效 | 鎮靜作用 |
| 唑尼沙胺 | 可能有效 | — |
| 吡拉西坦 | 可能有效 | — |
| 丙戊酸 | — | 原則禁忌(粒線體毒性) |
| 苯妥英 | 肌陣攣惡化 | 避免使用 |
| 卡巴馬西平 | 肌躍動惡化 | 避免使用 |
為輔助粒線體功能,以下補充劑經驗上被推薦使用,但缺乏強力證據1)。
建议进行物理治疗和有氧运动以维持运动能力。作业治疗和语言治疗也有帮助4)。
需要神經內科、眼科、心臟內科、內分泌內科的定期追蹤1)。建議每3個月進行臨床評估、血液檢查、腦電圖(EEG)及抗癲癇藥物血中濃度監測5)。
實驗證實丙戊酸會降低粒線體呼吸鏈複合體I和IV的活性,在MERRF這類粒線體疾病中可能惡化病情5)。癲癇治療應優先選用左乙拉西坦或氯硝西泮等粒線體毒性較低的藥物。
MERRF的病理源於mtDNA突變導致的粒線體功能障礙。
MT-TK基因的m.8344A>G突變會改變tRNA離胺酸的結構,進而干擾粒線體內的蛋白質合成(氧化磷酸化複合體的組裝)1)。結果導致ATP生成減少,能量需求高的器官(腦、骨骼肌、心肌)優先受損3)。
粒線體DNA突變以異質性狀態存在,當突變型mtDNA的比例超過組織特異性閾值時,會出現生化表現型3)。
作為對COX缺乏(呼吸鏈複合體IV障礙)的代償反應,肌纖維膜下會增生異常粒線體,形成破碎紅纖維(RRF)2)。電子顯微鏡下可觀察到異常延長的粒線體顯著增生以及類結晶包涵體2)。
視網膜神經節細胞能量需求高,對粒線體功能障礙較為脆弱1)。即使視覺上無症狀的患者,也可檢測到RNFL及GCC變薄,顯示潛在的神經細胞流失正在進行。
粒線體功能障礙會導致氧化壓力增加、免疫系統異常及粒線體動態改變,並參與癲癇發作的病理機制4)。
Baysal等人(2025)對一名攜帶m.8344A>G突變的38歲女性超難治性癲癇重積(SRSE)患者,植入了VNS裝置並採用快速遞增法(6天內增加至2 mA)4)。VNS植入7天後,肌陣攣開始改善,21天後全身性強直陣攣發作消失。在2年的隨訪中,全身性強直陣攣發作和癲癇重積未復發,肌陣攣也減少。VNS的作用機制被認為是通過刺激左側迷走神經,激活孤束核→藍斑核(釋放去甲腎上腺素)和中縫核(釋放血清素)通路,並上調GABA-A受體密度4)。
根據系統性回顧,VNS終止SE的比率報告為74%(38例中28例),但可能存在報告偏倚4)。
Perampanel和Rufinamide已被報導對非粒線體性肌陣攣癲癇有效,預期可應用於MERRF5)。
其他替代療法包括阿特金斯飲食、糖皮質激素、大麻二酚(CBD)、N-乙醯半胱氨酸、腦深部刺激、經顱磁刺激(可降低30-40%的發作頻率)正在研究中5)。
Kawazoe等人(2022)報告了一名68歲日本女性,帶有MT-TC基因的同質性突變m.5820C>A2)。該突變位於tRNA半胱氨酸的胺基酸接受臂根部,經電腦模擬分析預測具有致病性。該病例的皮膚切片發現p62陽性核內包涵體,顯示粒線體疾病與核內包涵體之間的新關聯2)。
這是一種將電極纏繞在左頸部迷走神經上,並透過植入胸部皮下的刺激裝置定期發送電刺激的治療方法。它經由腦幹發揮抗癲癇作用。曾有病例報告顯示對MERRF的超難治性癲癇重積狀態有效4),但證據有限。