L’epilessia mioclonica con fibre rosse sfilacciate (Myoclonic Epilepsy with Ragged Red Fibers: MERRF) è una rara malattia mitocondriale multisistemica causata da mutazioni del DNA mitocondriale. Nell’ICD-10 è classificata come E88.4 (disturbo del metabolismo mitocondriale).
Circa l’80% dei casi di MERRF è causato dalla mutazione puntiforme m.8344A>G nel gene MT-TK5). Questa mutazione provoca una disfunzione del tRNA della lisina, compromettendo l’assemblaggio delle proteine necessarie per la fosforilazione ossidativa1). Altre mutazioni causative riportate includono m.8356T>C, m.8363G>A e m.3243A>G5). È stata anche descritta una nuova mutazione nel gene MT-TC (m.5820C>A)2).
Nei casi ereditari, la trasmissione è quasi esclusivamente materna. Le principali malattie rappresentative delle encefalomiopatie mitocondriali sono tre: oftalmoplegia esterna progressiva cronica (CPEO), MELAS e MERRF, con MELAS che è la più frequente.
La prevalenza di MERRF è stimata in meno di 1 caso ogni 100.000 persone. L’età media di insorgenza è di 45 anni5), ma esistono casi con esordio precoce nell’infanzia con crisi epilettiche. È stato riportato che il 92,3% dei portatori della mutazione m.8344A>G sviluppa epilessia4).
Il fenotipo clinico varia notevolmente in base al grado di eteroplasmia (rapporto tra mtDNA mutato e wild-type nella stessa cellula)1)3). Con una bassa carica mutazionale possono verificarsi forme lievi senza sintomi del sistema nervoso centrale. Sono state descritte famiglie che non hanno manifestato sintomi neurologici per quattro generazioni3).
QCome viene ereditata la MERRF?
A
Il DNA mitocondriale viene ereditato solo dalla madre, quindi segue un modello di ereditarietà materna. La percentuale di mtDNA mutato (eteroplasmia) varia tra individui e tessuti, influenzando la gravità dei sintomi clinici. Per maggiori dettagli, vedere la sezione “Cause e fattori di rischio”.
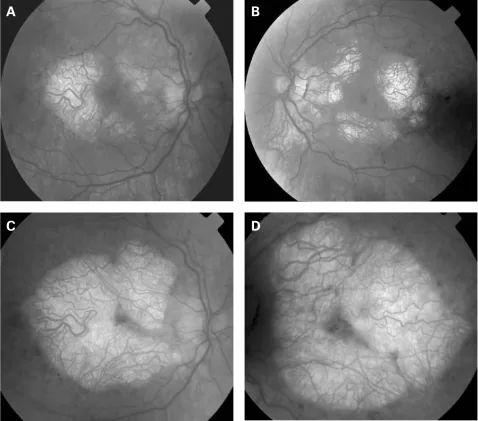
P P Rath, S Jenkins, M Michaelides et al. Characterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence. The British Journal of Ophthalmology. 2008 Jan 22; 92(5):623. Figure 2. PMCID: PMC2569141. License: CC BY.
La tetrade classica della MERRF comprende mioclono, epilessia generalizzata, atassia e fibre rosse sfilacciate alla biopsia muscolare. In ambito neuro-oftalmologico, i seguenti sintomi sono i principali disturbi.
Riduzione dell’acuità visiva: si verifica progressivamente a causa dell’atrofia ottica. Spesso è bilaterale e simmetrica.
Mioclono: contrazioni e rilasciamenti muscolari rapidi involontari, che di solito durano 1-2 secondi 5). Può essere scatenato dalla stimolazione luminosa 2).
Crisi epilettiche: le crisi miocloniche sono le più frequenti, ma sono state riportate anche crisi tonico-cloniche generalizzate, crisi focali e crisi di assenza5).
Atassia cerebellare: atassia e disartria compaiono progressivamente4).
Ipoacusia neurosensoriale: si associa con elevata frequenza.
Intorpidimento periferico: disturbi sensoriali distali dovuti a neuropatia periferica2).
Debolezza muscolare e facile affaticabilità: dovuti a miopatia4).
I principali reperti neuro-oftalmologici associati alla MERRF sono cinque: atrofia/neuropatia ottica, oftalmoplegia, ptosi palpebrale, retinopatia pigmentosa e nistagmo.
Atrofia ottica
Frequenza: 39% dei pazienti con MERRF (Hirano et al., 13/36 casi), 10% in tutti i portatori della mutazione m.8344A>G (Altmann et al., 34 casi) 1)
Reperti: pallore bilaterale e simmetrico della papilla ottica 1). In uno studio, l’atrofia ottica è stata riscontrata in 6 su 7 (85,7%) bambini e giovani adulti geneticamente confermati.
Punti di attenzione: in tutti e 3 i pazienti visivamente asintomatici è stato rilevato un assottigliamento dello strato di fibre nervose retiniche (RNFL) e dello strato di cellule gangliari maculari (GCC).
Oftalmoplegia e ptosi palpebrale
Oftalmoplegia: 2 su 34 casi (5,9%). La limitazione dei movimenti oculari estrinseci varia tra i report: 2/2 casi (Zhu et al.) e 3 su 7 casi (42,9%, Grönlund et al.).
Ptosi palpebrale: 10 su 34 casi (29,4%, Mancuso et al.). 1 su 7 casi (Grönlund et al.).
Retinopatia e nistagmo
Retinopatia pigmentosa: 4 su 24 casi (16,7%, Mancuso et al.). Distrofia retinica in 1 su 7 casi (Grönlund et al.).
QCi sono anomalie del nervo ottico anche se la vista è normale?
A
Anche in pazienti visivamente asintomatici, l’OCT può rilevare un assottigliamento dello strato di fibre nervose retiniche e del complesso di cellule gangliari maculari. Ciò riflette una perdita subclinica di cellule retiniche, pertanto si raccomanda un follow-up oculistico regolare.
Fibre rosse sfilacciate: presenti nel 96% dei casi con mutazione m.8344A>G alla biopsia muscolare1).
Lipomi multipli: prevalenza variabile dal 3% al 32,4% a seconda degli studi1). È stata segnalata un’associazione tra elevata eteroplasmia e dimensione dei lipomi1).
Cardiomiopatia, diabete e ipotiroidismo: si manifestano come parte del coinvolgimento multiorgano5).
Acidosi lattica: si osserva un aumento del lattato nel liquido cerebrospinale2).
Disabilità intellettiva e demenza: compaiono nei casi avanzati.
La causa della MERRF è una mutazione del DNA mitocondriale (mtDNA).
Mutazione m.8344A>G nel gene MT-TK: è la più frequente, rappresentando circa l’80% di tutti i casi5). Causa un’anomalia strutturale del tRNA della lisina, compromettendo la sintesi delle proteine necessarie per la fosforilazione ossidativa1).
Altre mutazioni del mtDNA: m.8356T>C, m.8363G>A, m.3243A>G, m.3255G>A, m.5820C>A nel gene MT-TC, ecc.2)5).
Ereditarietà materna: poiché il mtDNA viene ereditato solo dalla madre, nelle famiglie affette si trasmette solo ai discendenti per via materna.
Eteroplasmia ed effetto soglia: quando la percentuale di mtDNA mutante supera la soglia specifica del tessuto, compaiono anomalie biochimiche 3). La soglia nel muscolo scheletrico è stimata tra il 60% e il 90% di mutazione; se circa il 15% di mtDNA wild-type rimane, la traduzione e l’attività della COX si ripristinano quasi completamente 3).
Geni modificatori nucleari: si ipotizza l’esistenza di fattori che influenzano la segregazione e la replicazione del mtDNA, regolando la distribuzione della quantità di mutazione tra i tessuti 3).
Con la colorazione tricromica di Gomori modificata si confermano le fibre rosse sfilacciate. Si osservano fibre con forte colorazione SDH e fibre con deficit di COX. Con la doppia colorazione COX-SDH, circa il 5% delle fibre mostra deficit di COX 2). Al microscopio elettronico si osserva una proliferazione e un allungamento anomalo dei mitocondri sotto il sarcolemma 2).
Tomografia a coerenza ottica (OCT): è possibile rilevare l’assottigliamento dello strato di fibre nervose retiniche e del complesso delle cellule gangliari retiniche anche in pazienti asintomatici1)
Esame del fondo oculare: pallore della papilla ottica1)
Nelle fasi iniziali viene spesso erroneamente diagnosticata come JME, e la progressione di sintomi sistemici come atassia e declino cognitivo è utile per la diagnosi differenziale della PME1).
Attualmente non esiste una terapia curativa per la MERRF. Il trattamento è principalmente sintomatico e si basa su un approccio multidisciplinare che combina la gestione dell’epilessia e la terapia di supporto sistemica.
Per l’epilessia mioclonica, il levetiracetam è considerato il più efficace 5).
Terapia combinata levetiracetam + clonazepam: in uno studio su 17 pazienti con MERRF, tutti i 12 pazienti che sono passati dalla monoterapia alla terapia combinata hanno mostrato miglioramenti. Sono stati riportati anche miglioramenti della funzione cognitiva e della coordinazione motoria 5).
Clonazepam: come benzodiazepina, è efficace da solo per le mioclonie 5).
Zonisamide: è stata suggerita la sua efficacia 5).
Piracetam: efficace in alcuni casi 5). In un caso di una donna giapponese di 68 anni, dopo la sospensione di fenitoina e carbamazepina, il passaggio a una terapia a base di piracetam e levetiracetam ha migliorato i sintomi 2).
ASM
Effetto sulle mioclonie
Precauzioni
Levetiracetam
Efficace
Prima scelta
Clonazepam
Efficace
Sedazione
Zonisamide
Possibile efficacia
—
Piracetam
Possibile efficacia
—
Acido valproico
—
Controindicato in linea di principio (tossicità mitocondriale)
È necessario un follow-up regolare da parte di neurologia, oculistica, cardiologia e endocrinologia 1). Si raccomandano valutazioni cliniche, esami del sangue, EEG e monitoraggio dei livelli ematici di ASM ogni tre mesi 5).
QPerché l'acido valproico non può essere usato nella MERRF?
A
È stato dimostrato sperimentalmente che l’acido valproico riduce l’attività dei complessi I e IV della catena respiratoria mitocondriale, e nelle malattie mitocondriali come la MERRF può peggiorare la patologia 5). Nel trattamento dell’epilessia si preferiscono farmaci a bassa tossicità mitocondriale, come levetiracetam e clonazepam.
6. Fisiopatologia e meccanismi dettagliati di insorgenza
La mutazione m.8344A>G nel gene MT-TK altera la struttura del tRNA della lisina, compromettendo la sintesi proteica mitocondriale (assemblaggio dei complessi della fosforilazione ossidativa) 1). Di conseguenza, la produzione di ATP diminuisce e gli organi ad alto fabbisogno energetico (cervello, muscolo scheletrico, miocardio) vengono danneggiati preferenzialmente 3).
Le mutazioni del DNA mitocondriale sono presenti in stato di eteroplasmia e il fenotipo biochimico compare quando la percentuale di mtDNA mutato supera la soglia tessuto-specifica 3).
Soglia patologica nel muscolo scheletrico: 60-90% di mutazione 3)
Con circa il 15% di mtDNA wild-type residuo, la traduzione e l’attività della COX possono essere ripristinate a livelli quasi normali 3)
I geni modificatori nucleari regolano la segregazione e la replicazione del mtDNA in modo tessuto-specifico 3)
Come reazione compensatoria al deficit di COX (complesso IV della catena respiratoria), i mitocondri anomali proliferano sotto il sarcolemma, formando fibre rosse sfilacciate (RRF) 2). Al microscopio elettronico si osservano una marcata proliferazione di mitocondri anormalmente allungati e inclusioni paracristalline 2).
Le cellule gangliari retiniche hanno un elevato fabbisogno energetico e sono vulnerabili alla disfunzione mitocondriale 1). Anche in pazienti visivamente asintomatici, si rileva un assottigliamento di RNFL e GCC, indicando una progressiva perdita neuronale subclinica.
La disfunzione mitocondriale provoca un aumento dello stress ossidativo, anomalie del sistema immunitario e alterazioni della dinamica mitocondriale, contribuendo anche alla patogenesi delle crisi epilettiche 4).
7. Ricerche recenti e prospettive future (studi in fase di ricerca)
Baysal et al. (2025) hanno applicato la stimolazione rapida del nervo vago (VNS) con incremento rapido (fino a 2 mA in 6 giorni) in una donna di 38 anni con mutazione m.8344A>G e stato epilettico super-refrattario (SRSE) dopo impianto del dispositivo VNS4). Il miglioramento delle mioclonie è iniziato 7 giorni dopo l’impianto del VNS e le crisi tonico-cloniche generalizzate sono scomparse dopo 21 giorni. Durante un follow-up di 2 anni, non si sono verificate recidive di crisi tonico-cloniche generalizzate o stato epilettico, e le mioclonie sono diminuite. Come meccanismo d’azione del VNS, si ipotizza l’attivazione della via nucleo del tratto solitario → locus coeruleus (rilascio di noradrenalina) e nucleo del rafe (rilascio di serotonina) attraverso la stimolazione del nervo vago sinistro, nonché la regolazione verso l’alto della densità dei recettori GABA-A4).
Il tasso di cessazione delle SE con VNS è riportato in una revisione sistematica come 74% (28 su 38 casi), ma è stata segnalata la possibilità di un bias di pubblicazione4).
Perampanel e rufinamide hanno mostrato efficacia nelle mioclonie epilettiche non mitocondriali e si prevede la loro applicazione nella MERRF5).
Come altre terapie alternative, sono state studiate la dieta Atkins, i glucocorticoidi, il cannabidiolo (CBD), la N-acetilcisteina, la stimolazione cerebrale profonda e la stimolazione magnetica transcranica (riduzione della frequenza delle crisi del 30-40%)5).
Kawazoe et al. (2022) hanno riportato il caso di una donna giapponese di 68 anni con la mutazione omoplasmica m.5820C>A nel gene MT-TC2). Questa mutazione si trova alla base del braccio accettore dell’amminoacido del tRNA della cisteina ed è stata predetta come patogenica mediante analisi in silico. In questo caso, la biopsia cutanea ha mostrato inclusioni nucleari positive per p62, suggerendo una nuova associazione tra malattie mitocondriali e inclusioni nucleari2).
QCos'è la terapia di stimolazione del nervo vago?
A
È un trattamento che prevede l’avvolgimento di un elettrodo attorno al nervo vago nel collo sinistro e l’invio periodico di stimoli elettrici da un dispositivo impiantato sotto la pelle del torace. Esercita un effetto anticonvulsivante attraverso il tronco encefalico. Esiste un rapporto di caso in cui è stato efficace per lo stato epilettico super-refrattario nella MERRF4), ma le evidenze sono limitate.
Jeeva-Patel T, Freund P, Margolin EA. Lipomatosis and optic neuropathy clinches the diagnosis of myoclonic epilepsy with ragged red fibres (MERRF) syndrome. BMJ Case Rep. 2021;14:e240463.
Kawazoe T, Tobisawa S, Sugaya K, et al. Myoclonic epilepsy with ragged-red fibers with intranuclear inclusions. Intern Med. 2022;61:547-552.
Ripolone M, Zanotti S, Napoli L, et al. MERRF mutation A8344G in a four-generation family without central nervous system involvement: clinical and molecular characterization. J Pers Med. 2023;13:147.
Baysal L, Jobi S, Zimmermann S, Helmers A-K, Margraf NG. Successful application of vagus nerve stimulation in super refractory status epilepticus associated with MERRF syndrome. Epilepsy Behav Rep. 2025;30:100769. doi:10.1016/j.ebr.2025.100769.
Finsterer J. A review of the advances in the medical management of epilepsy associated with myoclonic epilepsy with ragged-red fibers (MERRF) syndrome. Cureus. 2025;17(4):e82875.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.