視神経萎縮
頻度:MERRF患者の39%(Hiranoら13/36例)、m.8344A>G変異保因者全体では10%(Altmannら34例)1)
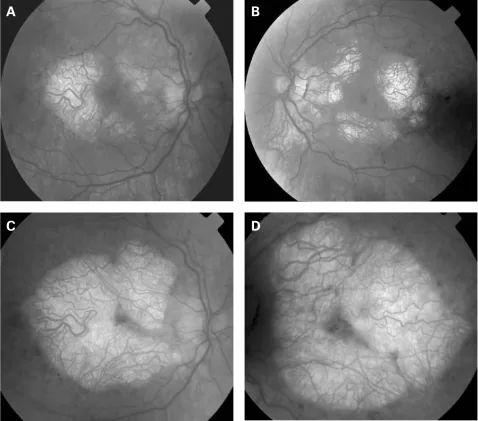
所見:両側性・対称的な視神経乳頭蒼白1)。遺伝学的に確認された小児・若年成人7例中6例(85.7%)で視神経萎縮を認めたとする報告もある。
注目点:視覚的に無症候な患者3例全例でRNFL・黄斑GCC菲薄化が検出された。

赤色ぼろ状線維を伴うミオクローヌスてんかん(Myoclonic Epilepsy with Ragged Red Fibers: MERRF)は、ミトコンドリアDNAの変異に起因する稀な多系統ミトコンドリア病である。ICD-10ではE88.4(ミトコンドリア代謝障害)に分類される。
MERRFの約80%はMT-TK遺伝子のm.8344A>G点変異により発症する5)。この変異はtRNAリシンの機能異常を引き起こし、酸化的リン酸化に必要なタンパク質の組み立てを障害する1)。その他の原因変異としてm.8356T>C、m.8363G>A、m.3243A>Gなどが報告されている5)。MT-TC遺伝子の新規変異(m.5820C>A)も報告されている2)。
遺伝性の場合、ほぼ母系遺伝の形式をとる。ミトコンドリア脳筋症の代表的疾患としては慢性進行性外眼筋麻痺(CPEO)、MELAS、MERRFの3つが挙げられ、MELASが最も多い。
MERRFの有病率は約10万人に1人以下と推定される。発症年齢の平均は45歳とされるが5)、幼児期早期にてんかん発作で発症する症例もある。m.8344A>G変異保因者の92.3%がてんかんを発症するとの報告がある4)。
ヘテロプラスミー(同一細胞内の変異型と野生型mtDNAの混在比率)の程度により臨床表現は大きく異なる1)3)。変異量が低い場合は中枢神経系症状を欠く軽症型もありうる。4世代にわたり中枢神経系の臨床症状を呈さなかった家系も報告されている3)。
ミトコンドリアDNAは母親からのみ受け継がれるため、母系遺伝の形式をとる。変異型mtDNAの割合(ヘテロプラスミー)は個人・組織ごとに異なり、臨床症状の重症度を左右する。詳細は「原因とリスク要因」の項を参照。
MERRFの古典的4徴はミオクローヌス、全般てんかん、失調、筋生検での赤色ぼろ状線維である。神経眼科領域では以下の症状が主訴となる。
MERRFに伴う主要な神経眼科的所見は視神経萎縮/視神経症、眼筋麻痺、眼瞼下垂、色素性網膜症、眼振の5つである。
視神経萎縮
頻度:MERRF患者の39%(Hiranoら13/36例)、m.8344A>G変異保因者全体では10%(Altmannら34例)1)
所見:両側性・対称的な視神経乳頭蒼白1)。遺伝学的に確認された小児・若年成人7例中6例(85.7%)で視神経萎縮を認めたとする報告もある。
注目点:視覚的に無症候な患者3例全例でRNFL・黄斑GCC菲薄化が検出された。
眼筋麻痺・眼瞼下垂
網膜症・眼振
色素性網膜症:24例中4例(16.7%、Mancusoら)。7例中1例の網膜ジストロフィー(Grönlundら)。
眼振:7例中1例(Grönlundら)。
視覚的に無症候な患者でもOCT検査で網膜神経線維層や黄斑部神経節細胞複合体の菲薄化が検出される。潜在的な網膜神経細胞の脱落を反映しており、定期的な眼科フォローが推奨される。
MERRFの原因はミトコンドリアDNA(mtDNA)の変異である。
MERRFの診断には以下の4つの臨床的特徴(古典的4徴)が用いられる。
確定診断には分子遺伝学的検査が必須である。患者の80%以上でm.8344A>G変異が同定される5)。
Gomoriのtrichrome変法で赤色ぼろ状線維を確認する。SDH強染色、COX欠損線維を認める。COX-SDH二重染色ではCOX欠損線維が約5%にみられる2)。電子顕微鏡では筋鞘膜下のミトコンドリア異常増殖・伸長が観察される2)。
乳酸19 mg/dL(正常4.2〜17.0)、ピルビン酸1.2 mg/dL(正常0.3〜0.9)の軽度上昇2)。
骨格筋のcomplex IV活性が対照の55〜69%に低下2)。
MERRFは進行性ミオクローヌスてんかん(PME)の一型であり、以下との鑑別が重要である。
| 疾患 | 鑑別のポイント |
|---|---|
| 若年性ミオクローヌスてんかん(JME) | 認知機能低下・失調の進行がなく、予後良好 |
| Lafora病 | 皮膚生検でLafora小体陽性 |
| Unverricht-Lundborg病 | CSTB遺伝子変異、進行が緩徐 |
| 神経セロイドリポフスチン症 | 網膜変性が顕著、酵素欠損 |
初期にはJMEと誤診されやすく、失調や認知機能低下の進行など全身性の随伴症状がPMEの鑑別に有用である1)。
MERRFに対する根治療法は現時点で存在しない。治療は対症療法が中心であり、てんかん管理と全身支持療法を組み合わせた多診療科的アプローチが基本となる。
ミオクローヌスてんかんに対してはレベチラセタムが最も有効とされる5)。
| ASM | ミオクローヌスへの効果 | 注意事項 |
|---|---|---|
| レベチラセタム | 有効 | 第一選択 |
| クロナゼパム | 有効 | 鎮静作用 |
| ゾニサミド | 有効の可能性 | — |
| ピラセタム | 有効の可能性 | — |
| バルプロ酸 | — | 原則禁忌(ミトコンドリア毒性) |
| フェニトイン | ミオクローヌス悪化 | 使用回避 |
| カルバマゼピン | ミオクローヌス悪化 | 使用回避 |
ミトコンドリア機能を補助する目的で以下のサプリメントが経験的に推奨されるが、強いエビデンスはない1)。
理学療法、有酸素運動による運動能力の維持が推奨される。作業療法および言語療法も有用である4)。
神経内科、眼科、循環器内科、内分泌内科の定期的フォローが必要である1)。3か月ごとの臨床評価、血液検査、EEG、ASM血中濃度モニタリングが推奨される5)。
バルプロ酸はミトコンドリア呼吸鎖complex IおよびIVの活性を低下させることが実験的に確認されており、MERRFのようなミトコンドリア病では病態を悪化させる危険性がある5)。てんかん治療ではレベチラセタムやクロナゼパムなど、ミトコンドリア毒性が低い薬剤が優先される。
MERRFの病態はmtDNA変異によるミトコンドリア機能障害に起因する。
MT-TK遺伝子のm.8344A>G変異はtRNAリシンの構造を変化させ、ミトコンドリア内でのタンパク質合成(酸化的リン酸化複合体の組み立て)を障害する1)。結果としてATP産生が低下し、エネルギー需要の高い臓器(脳、骨格筋、心筋)が優先的に障害を受ける3)。
ミトコンドリアDNA変異はヘテロプラスミーの状態で存在し、変異型mtDNAの割合が組織特異的な閾値を超えると生化学的表現型が出現する3)。
COX欠損(呼吸鎖complex IV障害)に対する代償反応として、筋鞘膜下に異常ミトコンドリアが増殖し、赤色ぼろ状線維(RRF)を形成する2)。電子顕微鏡では異常に伸長したミトコンドリアの著明な増殖と、パラクリスタリン封入体が観察される2)。
網膜神経節細胞はエネルギー需要が高く、ミトコンドリア機能障害に対して脆弱である1)。視覚的に無症候な患者でもRNFL・GCCの菲薄化が検出されており、潜在的な神経細胞脱落が進行していると考えられる。
ミトコンドリア機能障害は酸化ストレスの増加、免疫系の異常、ミトコンドリア動態の変容を引き起こし、てんかん発作の病態にも関与する4)。
Baysalら(2025)は、m.8344A>G変異を持つ38歳女性の超難治性てんかん重積(SRSE)に対し、VNSデバイスを植え込み急速漸増法(6日間で2 mAまで増量)を適用した4)。VNS植え込み7日後にミオクローヌスの改善が始まり、21日後には全般性強直間代発作が消失した。2年間のフォローアップで全般性強直間代発作・てんかん重積の再発はなく、ミオクローヌスも減少した。VNSの作用機序として、左迷走神経刺激を介した孤束核→青斑核(ノルエピネフリン放出)・縫線核(セロトニン放出)経路の活性化およびGABA-A受容体密度の上方制御が推定されている4)。
VNSによるSE停止率は系統的レビューで74%(38例中28例)と報告されているが、報告バイアスの可能性が指摘されている4)。
ペランパネルおよびルフィナマイドは非ミトコンドリア性のミオクローヌスてんかんで有効性が報告されており、MERRFへの応用が期待される5)。
その他の代替療法として、アトキンス食、グルココルチコイド、カンナビジオール(CBD)、N-アセチルシステイン、脳深部刺激、経頭蓋磁気刺激(30〜40%の発作頻度低下)が検討されている5)。
Kawazoeら(2022)は、MT-TC遺伝子のホモプラスミー変異m.5820C>Aを有する68歳日本人女性を報告した2)。本変異はtRNAシステインのアミノ酸受容体ステムの根元に位置し、in silico解析で病原性が予測された。この症例では皮膚生検でp62陽性核内封入体が認められ、ミトコンドリア疾患と核内封入体の関連という新たな知見が示された2)。
左頸部の迷走神経に電極を巻きつけ、胸部皮下に植え込んだ刺激装置から周期的に電気刺激を送る治療法である。脳幹を経由して抗けいれん作用を発揮する。MERRFの超難治性てんかん重積に対して有効であった症例報告がある4)が、エビデンスは限定的である。