Myoclonic Epilepsy with Ragged Red Fibers (MERRF) is a rare multisystem mitochondrial disease caused by mutations in mitochondrial DNA. It is classified under ICD-10 code E88.4 (mitochondrial metabolism disorder).
Approximately 80% of MERRF cases are caused by the m.8344A>G point mutation in the MT-TK gene 5). This mutation leads to dysfunction of tRNA lysine, impairing the assembly of proteins required for oxidative phosphorylation 1). Other causative mutations include m.8356T>C, m.8363G>A, and m.3243A>G 5). A novel mutation in the MT-TC gene (m.5820C>A) has also been reported 2).
Inherited cases are almost exclusively maternally inherited. The three major representative mitochondrial encephalomyopathies are chronic progressive external ophthalmoplegia (CPEO), MELAS, and MERRF, with MELAS being the most common.
The prevalence of MERRF is estimated to be less than 1 in 100,000 people. The average age of onset is reported to be 45 years5), but some cases present with epileptic seizures in early childhood. It has been reported that 92.3% of carriers of the m.8344A>G mutation develop epilepsy4).
Clinical manifestations vary greatly depending on the degree of heteroplasmy (the proportion of mutant and wild-type mtDNA within the same cell)1)3). When the mutation load is low, mild forms lacking central nervous system symptoms may occur. Families spanning four generations without central nervous system clinical symptoms have also been reported3).
QHow is MERRF inherited?
A
Since mitochondrial DNA is inherited only from the mother, MERRF follows a maternal inheritance pattern. The proportion of mutant mtDNA (heteroplasmy) varies among individuals and tissues, influencing the severity of clinical symptoms. For details, see the “Causes and Risk Factors” section.
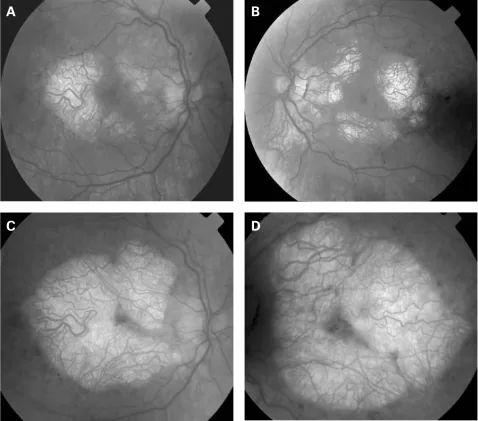
P P Rath, S Jenkins, M Michaelides et al. Characterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence. The British Journal of Ophthalmology. 2008 Jan 22; 92(5):623. Figure 2. PMCID: PMC2569141. License: CC BY.
A 1994 photograph of a patient with MERRF syndrome showing multiple lipomas on the trunk and upper limbs
The classic tetrad of MERRF includes myoclonus, generalized epilepsy, ataxia, and ragged-red fibers on muscle biopsy. In the neuro-ophthalmology field, the following symptoms are the main complaints.
Vision loss: Progressively occurs with optic atrophy. Often bilateral and symmetric.
Myoclonus: Involuntary rapid muscle contraction and relaxation, usually lasting 1–2 seconds 5). May be triggered by light stimulation 2).
Epileptic seizures: Myoclonic seizures are most common, but generalized tonic-clonic seizures, focal seizures, and absence seizures have also been reported 5).
Cerebellar ataxia: Progressive motor ataxia and dysarthria appear 4).
The main neuro-ophthalmological findings associated with MERRF are optic atrophy/optic neuropathy, ophthalmoplegia, ptosis, pigmentary retinopathy, and nystagmus.
Optic Atrophy
Frequency: 39% of MERRF patients (Hirano et al. 13/36 cases), 10% overall in carriers of the m.8344A>G mutation (Altmann et al. 34 cases)1)
Findings: Bilateral, symmetric optic disc pallor1). One report noted optic atrophy in 6 of 7 (85.7%) genetically confirmed children and young adults.
Key point: RNFL and macular GCC thinning was detected in all 3 visually asymptomatic patients.
Ophthalmoplegia and Ptosis
Ophthalmoplegia: 2 of 34 cases (5.9%). Reports vary: extraocular muscle restriction in 2/2 cases (Zhu et al.) and 3 of 7 cases (42.9%, Grönlund et al.).
Ptosis: 10 out of 34 cases (29.4%, Mancuso et al.). 1 out of 7 cases (Grönlund et al.).
Retinopathy and nystagmus
Pigmentary retinopathy: 4 out of 24 cases (16.7%, Mancuso et al.). Retinal dystrophy in 1 out of 7 cases (Grönlund et al.).
QCan the optic nerve be abnormal even if visual acuity is normal?
A
Even in visually asymptomatic patients, OCT can detect thinning of the retinal nerve fiber layer and macular ganglion cell complex. This reflects potential loss of retinal nerve cells, and regular ophthalmologic follow-up is recommended.
Ragged-red fibers: Confirmed by muscle biopsy in 96% of cases with the m.8344A>G mutation1).
Multiple lipomas: Prevalence ranges from 3% to 32.4% depending on the report1). An association between high heteroplasmy and lipoma size has been noted1).
Cardiomyopathy, diabetes, hypothyroidism: These occur as part of multi-organ involvement5).
Lactic acidosis: Elevated cerebrospinal fluid lactate levels are observed2).
Intellectual disability/dementia: Appears in advanced cases.
MERRF is caused by mutations in mitochondrial DNA (mtDNA).
m.8344A>G mutation in the MT-TK gene: Most common, accounting for about 80% of all cases5). It causes structural abnormalities in tRNA lysine, impairing the synthesis of proteins required for oxidative phosphorylation1).
Other mtDNA mutations: m.8356T>C, m.8363G>A, m.3243A>G, m.3255G>A, m.5820C>A in the MT-TC gene, etc.2)5).
Maternal inheritance: Since mtDNA is inherited only from the mother, it is transmitted only through the maternal line in affected families.
Heteroplasmy and threshold effect: When the proportion of mutant mtDNA exceeds a tissue-specific threshold, biochemical abnormalities appear3). The threshold in skeletal muscle is estimated to be 60–90% mutant load, and if about 15% wild-type mtDNA remains, translation and COX activity can be almost normal3).
Nuclear modifier genes: Factors that influence mtDNA segregation and replication, regulating the distribution of mutation load among tissues, have been suggested3).
Gomori’s modified trichrome staining reveals ragged-red fibers. SDH hyperstaining and COX-deficient fibers are observed. In COX-SDH double staining, COX-deficient fibers are seen in about 5% of cases2). Electron microscopy shows abnormal proliferation and elongation of mitochondria beneath the sarcolemma2).
Optical Coherence Tomography (OCT): Can detect thinning of the retinal nerve fiber layer and retinal ganglion cell complex even in asymptomatic patients1)
In the early stages, it is often misdiagnosed as JME, and systemic accompanying symptoms such as ataxia and progression of cognitive decline are useful for differentiating PME1).
Currently, there is no curative treatment for MERRF. Treatment is primarily symptomatic, and a multidisciplinary approach combining epilepsy management with systemic supportive care is fundamental.
Levetiracetam is considered the most effective treatment for myoclonic epilepsy 5).
Levetiracetam + clonazepam combination therapy: In a study of 17 patients with MERRF, all 12 patients who switched from monotherapy to combination therapy showed improvement. Improvements in cognitive function and coordination have also been reported 5).
Clonazepam: As a benzodiazepine, it is effective for myoclonus even when used alone5).
Zonisamide: Efficacy has been suggested5).
Piracetam: Effective in some cases5). In a case of a 68-year-old Japanese woman, symptoms improved after switching from phenytoin and carbamazepine to a regimen primarily consisting of piracetam and levetiracetam2).
ASM
Effect on myoclonus
Precautions
Levetiracetam
Effective
First-line
Clonazepam
Effective
Sedation
Zonisamide
Possibly effective
—
Piracetam
Possibly effective
—
Valproic acid
—
Generally contraindicated (mitochondrial toxicity)
Regular follow-up by neurology, ophthalmology, cardiology, and endocrinology is necessary1). Clinical evaluation, blood tests, EEG, and ASM blood level monitoring every three months are recommended5).
QWhy can't valproate be used for MERRF?
A
Valproate has been experimentally confirmed to reduce the activity of mitochondrial respiratory chain complexes I and IV, and may worsen the condition in mitochondrial diseases such as MERRF5). For epilepsy treatment, drugs with low mitochondrial toxicity, such as levetiracetam and clonazepam, are preferred.
The m.8344A>G mutation in the MT-TK gene alters the structure of tRNA lysine, impairing protein synthesis (assembly of oxidative phosphorylation complexes) within mitochondria 1). As a result, ATP production decreases, and organs with high energy demands (brain, skeletal muscle, heart) are preferentially affected 3).
Mitochondrial DNA mutations exist in a state of heteroplasmy, and when the proportion of mutant mtDNA exceeds a tissue-specific threshold, a biochemical phenotype appears3).
Pathological threshold in skeletal muscle: 60–90% mutant load3)
With approximately 15% residual wild-type mtDNA, translation and COX activity can be restored to nearly normal levels3)
Nuclear modifier genes regulate mtDNA segregation and replication in a tissue-specific manner3)
As a compensatory response to COX deficiency (respiratory chain complex IV impairment), abnormal mitochondria proliferate beneath the sarcolemma, forming ragged red fibers (RRFs) 2). Electron microscopy reveals marked proliferation of abnormally elongated mitochondria and paracrystalline inclusions 2).
Retinal ganglion cells have high energy demands and are vulnerable to mitochondrial dysfunction 1). Even in visually asymptomatic patients, thinning of the RNFL and GCC is detected, suggesting ongoing subclinical neuronal loss.
Mitochondrial dysfunction causes increased oxidative stress, immune system abnormalities, and alterations in mitochondrial dynamics, and is also involved in the pathology of epileptic seizures4).
7. Latest Research and Future Perspectives (Reports at the Research Stage)
Baysal et al. (2025) implanted a VNS device and applied a rapid titration method (increasing to 2 mA over 6 days) in a 38-year-old woman with the m.8344A>G mutation who had super-refractory status epilepticus (SRSE)4). Improvement in myoclonus began 7 days after VNS implantation, and generalized tonic-clonic seizures disappeared after 21 days. During a 2-year follow-up, there was no recurrence of generalized tonic-clonic seizures or status epilepticus, and myoclonus also decreased. The mechanism of action of VNS is presumed to involve activation of the nucleus tractus solitarius→locus coeruleus (norepinephrine release) and raphe nuclei (serotonin release) pathways via left vagus nerve stimulation, as well as upregulation of GABA-A receptor density4).
The seizure termination rate with VNS is reported as 74% (28 out of 38 cases) in a systematic review, but the possibility of reporting bias has been pointed out4).
Perampanel and rufinamide have been reported to be effective in non-mitochondrial myoclonus epilepsy, and their application to MERRF is expected5).
Other alternative therapies being investigated include the Atkins diet, glucocorticoids, cannabidiol (CBD), N-acetylcysteine, deep brain stimulation, and transcranial magnetic stimulation (30–40% reduction in seizure frequency)5).
Kawazoe et al. (2022) reported a 68-year-old Japanese woman with a homoplasmic mutation m.5820C>A in the MT-TC gene 2). This mutation is located at the base of the amino acid acceptor stem of tRNA cysteine, and in silico analysis predicted its pathogenicity. In this case, skin biopsy revealed p62-positive intranuclear inclusions, providing new insights into the association between mitochondrial disease and intranuclear inclusions 2).
QWhat is vagus nerve stimulation therapy?
A
This treatment involves wrapping electrodes around the left vagus nerve in the neck and periodically delivering electrical stimulation from a device implanted under the chest skin. It exerts anticonvulsant effects via the brainstem. There have been case reports of its effectiveness for super-refractory status epilepticus in MERRF 4), but evidence is limited.
Jeeva-Patel T, Freund P, Margolin EA. Lipomatosis and optic neuropathy clinches the diagnosis of myoclonic epilepsy with ragged red fibres (MERRF) syndrome. BMJ Case Rep. 2021;14:e240463.
Kawazoe T, Tobisawa S, Sugaya K, et al. Myoclonic epilepsy with ragged-red fibers with intranuclear inclusions. Intern Med. 2022;61:547-552.
Ripolone M, Zanotti S, Napoli L, et al. MERRF mutation A8344G in a four-generation family without central nervous system involvement: clinical and molecular characterization. J Pers Med. 2023;13:147.
Baysal L, Jobi S, Zimmermann S, Helmers A-K, Margraf NG. Successful application of vagus nerve stimulation in super refractory status epilepticus associated with MERRF syndrome. Epilepsy Behav Rep. 2025;30:100769. doi:10.1016/j.ebr.2025.100769.
Finsterer J. A review of the advances in the medical management of epilepsy associated with myoclonic epilepsy with ragged-red fibers (MERRF) syndrome. Cureus. 2025;17(4):e82875.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.