Die Myoklonus-Epilepsie mit Ragged Red Fibers (MERRF) ist eine seltene, durch Mutationen der mitochondrialen DNA verursachte Multisystem-Mitochondrienerkrankung. In der ICD-10 wird sie unter E88.4 (Mitochondriale Stoffwechselstörung) klassifiziert.
Etwa 80 % der MERRF-Fälle werden durch die Punktmutation m.8344A>G im MT-TK-Gen verursacht 5). Diese Mutation führt zu einer Funktionsstörung der tRNA-Lysin und beeinträchtigt den Zusammenbau von Proteinen, die für die oxidative Phosphorylierung benötigt werden 1). Weitere ursächliche Mutationen sind m.8356T>C, m.8363G>A und m.3243A>G 5). Auch eine neue Mutation im MT-TC-Gen (m.5820C>A) wurde berichtet 2).
Bei erblichen Fällen erfolgt die Vererbung fast ausschließlich mütterlicherseits. Zu den typischen mitochondrialen Enzephalomyopathien zählen die chronisch progressive externe Ophthalmoplegie (CPEO), MELAS und MERRF, wobei MELAS am häufigsten ist.
Die Prävalenz von MERRF wird auf weniger als 1 pro 100.000 Personen geschätzt. Das durchschnittliche Erkrankungsalter liegt bei 45 Jahren 5), es gibt jedoch auch Fälle mit frühem Beginn im Kindesalter mit epileptischen Anfällen. Berichten zufolge entwickeln 92,3 % der Träger der m.8344A>G-Mutation Epilepsie 4).
Das klinische Erscheinungsbild variiert stark je nach Grad der Heteroplasmie (Mischungsverhältnis von mutierter und Wildtyp-mtDNA in derselben Zelle) 1)3). Bei geringer Mutationslast kann eine milde Form ohne Symptome des zentralen Nervensystems auftreten. Es wurden auch Familien über vier Generationen beschrieben, die keine klinischen Symptome des zentralen Nervensystems zeigten 3).
QWie wird MERRF vererbt?
A
Da die mitochondriale DNA nur von der Mutter vererbt wird, erfolgt die Vererbung mütterlicherseits. Der Anteil der mutierten mtDNA (Heteroplasmie) variiert zwischen Individuen und Geweben und beeinflusst die Schwere der klinischen Symptome. Details finden Sie im Abschnitt „Ursachen und Risikofaktoren“.
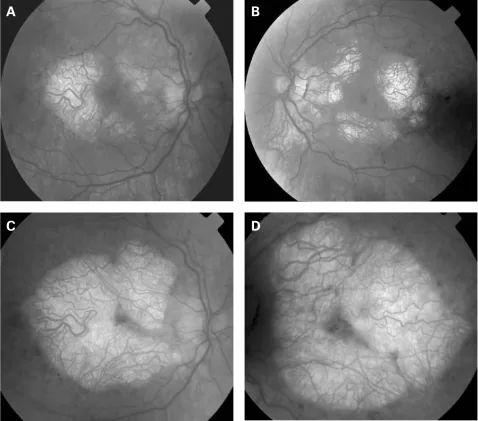
P P Rath, S Jenkins, M Michaelides et al. Characterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence. The British Journal of Ophthalmology. 2008 Jan 22; 92(5):623. Figure 2. PMCID: PMC2569141. License: CC BY.
Abbildung: Klinische Merkmale des MERRF-Syndroms (1994–2001)
Die klassische Tetrade des MERRF-Syndroms umfasst Myoklonus, generalisierte Epilepsie, Ataxie und rote zerfaserte Fasern in der Muskelbiopsie. Im Bereich der Neuroophthalmologie stehen folgende Symptome im Vordergrund.
Sehverschlechterung: Tritt fortschreitend mit Optikusatrophie auf. Meist bilateral und symmetrisch.
Myoklonus: Unwillkürliche, schnelle Muskelkontraktionen und -erschlaffungen, die normalerweise 1–2 Sekunden anhalten 5). Kann durch Lichtreize ausgelöst werden 2).
Epileptische Anfälle: Myoklonische Anfälle sind am häufigsten, aber auch generalisierte tonisch-klonische Anfälle, fokale Anfälle und Absencen wurden berichtet5).
Kleinhirnataxie: Ataxie und Dysarthrie treten fortschreitend auf4).
Schallempfindungsschwerhörigkeit: Tritt häufig auf.
Peripheres Taubheitsgefühl: Distal betonte sensorische Störungen aufgrund einer peripheren Neuropathie2).
Muskelschwäche und leichte Ermüdbarkeit: Aufgrund einer Myopathie4).
Die wichtigsten neuroophthalmologischen Befunde bei MERRF sind Optikusatrophie/Optikusneuropathie, Ophthalmoplegie, Ptosis, Pigmentretinopathie und Nystagmus.
Optikusatrophie
Häufigkeit: 39% der MERRF-Patienten (Hirano et al., 13/36 Fälle), insgesamt 10% der Träger der m.8344A>G-Mutation (Altmann et al., 34 Fälle) 1)
Befund: Bilaterale, symmetrische Blässe der Papille 1). In einer Studie wurde bei 6 von 7 (85,7%) genetisch bestätigten Kindern und jungen Erwachsenen eine Optikusatrophie festgestellt.
Besonderheit: Bei allen 3 visuell asymptomatischen Patienten wurde eine Ausdünnung der RNFL und des makulären GCC nachgewiesen.
Augenmuskellähmung und Ptosis
Augenmuskellähmung: 2 von 34 Fällen (5,9%). Die Einschränkung der äußeren Augenmuskelbeweglichkeit variiert zwischen den Berichten: 2/2 Fälle (Zhu et al.) und 3 von 7 Fällen (42,9%, Grönlund et al.).
Ptosis: 10 von 34 Fällen (29,4%, Mancuso et al.). 1 von 7 Fällen (Grönlund et al.).
Retinopathie und Nystagmus
Pigmentretinopathie: 4 von 24 Fällen (16,7%, Mancuso et al.). 1 von 7 Fällen mit Netzhautdystrophie (Grönlund et al.).
QGibt es Auffälligkeiten am Sehnerv, auch wenn das Sehvermögen normal ist?
A
Auch bei visuell asymptomatischen Patienten kann die OCT eine Ausdünnung der retinalen Nervenfaserschicht und des makulären Ganglienzellkomplexes nachweisen. Dies spiegelt einen potenziellen Verlust retinaler Nervenzellen wider, und regelmäßige augenärztliche Kontrollen werden empfohlen.
Rote zerfetzte Fasern: Bei 96% der Patienten mit m.8344A>G-Mutation durch Muskelbiopsie nachgewiesen 1).
Multiple Lipome: Die Häufigkeit variiert zwischen 3 und 32,4% je nach Studie 1). Ein Zusammenhang zwischen hoher Heteroplasmie und Lipomgröße wurde beschrieben 1).
Kardiomyopathie, Diabetes mellitus, Hypothyreose: treten als Teil einer Multiorganerkrankung auf 5).
Laktatazidose: Erhöhung der Laktatkonzentration im Liquor 2).
Intelligenzminderung/Demenz: treten in fortgeschrittenen Fällen auf.
Die Ursache des MERRF-Syndroms sind Mutationen der mitochondrialen DNA (mtDNA).
m.8344A>G-Mutation im MT-TK-Gen: die häufigste Mutation, die etwa 80% aller Fälle ausmacht 5). Sie führt zu einer strukturellen Anomalie der tRNA-Lysin und beeinträchtigt die Synthese von Proteinen, die für die oxidative Phosphorylierung notwendig sind 1).
Andere mtDNA-Mutationen: m.8356T>C, m.8363G>A, m.3243A>G, m.3255G>A, m.5820C>A im MT-TC-Gen u.a. 2)5).
Maternale Vererbung: Da mtDNA nur von der Mutter weitergegeben wird, wird die Erkrankung in betroffenen Familien nur über die mütterliche Linie vererbt.
Heteroplasmie und Schwellenwerteffekt: Wenn der Anteil der mutierten mtDNA einen gewebespezifischen Schwellenwert überschreitet, treten biochemische Anomalien auf 3). Der Schwellenwert im Skelettmuskel wird auf 60–90 % Mutationslast geschätzt; wenn etwa 15 % Wildtyp-mtDNA verbleiben, normalisieren sich Translation und COX-Aktivität nahezu vollständig 3).
Modifikatorgene im Zellkern: Es wird angenommen, dass es Faktoren gibt, die die Segregation und Replikation der mtDNA beeinflussen und die Verteilung der Mutationslast zwischen den Geweben regulieren 3).
Für die definitive Diagnose ist eine molekulargenetische Untersuchung erforderlich. Bei über 80 % der Patienten wird die Mutation m.8344A>G identifiziert 5).
Probenmaterial: DNA-Extraktion aus Blut, Urin oder Muskel
Methode: Quantifizierung der Heteroplasmie mittels PCR-RFLP 3)
Hinweis: In der Regel ist die Heteroplasmie im Urin höher als im Blut 3)
Mit der modifizierten Gomori-Trichrom-Färbung werden rote zerfaserte Fasern nachgewiesen. Es zeigen sich SDH-Hyperfärbung und COX-defiziente Fasern. In der COX-SDH-Doppelfärbung sind etwa 5% der Fasern COX-defizient 2). Elektronenmikroskopisch werden eine abnorme Vermehrung und Verlängerung der Mitochondrien unter dem Sarkolemm beobachtet 2).
Nervenleitungsuntersuchung: Zeigt ein Muster einer sensorischen Neuropathie 2)
Elektroenzephalographie (EEG): Es besteht eine Lichtempfindlichkeit. Charakteristisch sind riesige somatosensorisch evozierte Potenziale (SEP 90 μV, normal <10 μV) 2)
Elektromyographie: Myogene Veränderungen in den distalen unteren Extremitäten 2)
Magnetresonanztomographie des Gehirns (cMRT): Es können eine Kleinhirnatrophie und Signalveränderungen im Nucleus dentatus auftreten 2). Oft ist der Befund jedoch normal 1)
MR-Spektroskopie: Oft wird ein Laktat-Peak nachgewiesen 1)
Optische Kohärenztomographie (OCT): Eine Verdünnung der retinalen Nervenfaserschicht und des retinalen Ganglienzellkomplexes kann auch bei asymptomatischen Patienten nachgewiesen werden 1)
Anfangs wird sie leicht mit JME verwechselt, und systemische Begleitsymptome wie Ataxie und kognitiver Abbau sind nützlich für die Differenzialdiagnose der PME1).
Derzeit gibt es keine kurative Behandlung für MERRF. Die Behandlung konzentriert sich auf symptomatische Therapie, und ein multidisziplinärer Ansatz, der Epilepsiemanagement und allgemeine unterstützende Maßnahmen kombiniert, ist die Grundlage.
Levetiracetam gilt als am wirksamsten gegen Myoklonus-Epilepsie 5).
Levetiracetam + Clonazepam Kombinationstherapie: In einer Studie mit 17 MERRF-Patienten zeigten alle 12 Patienten, die von einer Monotherapie auf eine Kombinationstherapie umgestellt wurden, eine Verbesserung. Auch Verbesserungen der kognitiven Funktion und der Koordination wurden berichtet 5).
Clonazepam: Als Benzodiazepin allein wirksam gegen Myoklonus 5).
Zonisamid: Wirksamkeit wird vermutet 5).
Piracetam: Wirksam in einigen Fällen 5). Bei einer 68-jährigen Japanerin führte der Wechsel von Phenytoin und Carbamazepin zu einem Regime mit Piracetam und Levetiracetam zu einer Symptombesserung 2).
Regelmäßige Nachsorge in Neurologie, Augenheilkunde, Kardiologie und Endokrinologie ist erforderlich 1). Klinische Bewertung, Blutuntersuchungen, EEG und Überwachung der ASM-Blutspiegel werden alle drei Monate empfohlen 5).
QWarum kann Valproat bei MERRF nicht eingesetzt werden?
A
Es wurde experimentell bestätigt, dass Valproat die Aktivität der mitochondrialen Atmungskettenkomplexe I und IV verringert, was bei mitochondrialen Erkrankungen wie MERRF das Risiko einer Verschlechterung des Krankheitszustands birgt 5). Bei der Epilepsiebehandlung werden Medikamente mit geringer mitochondrialer Toxizität wie Levetiracetam oder Clonazepam bevorzugt.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Die m.8344A>G-Mutation im MT-TK-Gen verändert die Struktur der tRNA-Lysin und beeinträchtigt die Proteinsynthese (Zusammenbau der oxidativen Phosphorylierungskomplexe) in den Mitochondrien 1). Dies führt zu einer verminderten ATP-Produktion, wodurch Organe mit hohem Energiebedarf (Gehirn, Skelettmuskulatur, Herzmuskel) bevorzugt geschädigt werden 3).
Mitochondriale DNA-Mutationen liegen im Zustand der Heteroplasmie vor, und wenn der Anteil der mutierten mtDNA einen gewebespezifischen Schwellenwert überschreitet, tritt ein biochemischer Phänotyp auf 3).
Pathologischer Schwellenwert im Skelettmuskel: Mutationsanteil 60–90 % 3)
Mit etwa 15 % verbleibender Wildtyp-mtDNA können Translation und COX-Aktivität nahezu normale Werte wiederherstellen 3)
Nukleäre modifizierende Gene regulieren die Trennung und Replikation der mtDNA gewebespezifisch 3)
Als kompensatorische Reaktion auf den COX-Mangel (Atmungskettenkomplex-IV-Störung) vermehren sich abnorme Mitochondrien unter dem Sarkolemm und bilden ragged red fibers (RRF) 2). Elektronenmikroskopisch sind eine auffällige Vermehrung abnormal verlängerter Mitochondrien und parakristalline Einschlusskörperchen zu beobachten 2).
Retinale Ganglienzellen haben einen hohen Energiebedarf und sind anfällig für mitochondriale Funktionsstörungen 1). Auch bei visuell asymptomatischen Patienten werden eine Ausdünnung von RNFL und GCC nachgewiesen, was auf einen fortschreitenden neuronalen Zellverlust hindeutet.
Mitochondriale Dysfunktion führt zu erhöhtem oxidativem Stress, Anomalien des Immunsystems und Veränderungen der Mitochondriendynamik und ist auch an der Pathogenese von epileptischen Anfällen beteiligt4).
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Baysal et al. (2025) wandten bei einer 38-jährigen Frau mit m.8344A>G-Mutation und superrefraktärem Status epilepticus (SRSE) nach Implantation eines VNS-Geräts ein schnelles Titrationsschema (Steigerung auf 2 mA innerhalb von 6 Tagen) an 4). Sieben Tage nach der VNS-Implantation begann die Besserung der Myoklonien, und nach 21 Tagen verschwanden die generalisierten tonisch-klonischen Anfälle. Während des zweijährigen Follow-ups traten weder generalisierte tonisch-klonische Anfälle noch ein Status epilepticus erneut auf, und die Myoklonien nahmen ab. Als Wirkmechanismus von VNS wird die Aktivierung des Nucleus tractus solitarius über die Stimulation des linken Vagusnervs, gefolgt von der Aktivierung des Locus caeruleus (Noradrenalinfreisetzung) und des Nucleus raphe (Serotoninfreisetzung) sowie die Hochregulierung der GABA-A-Rezeptordichte angenommen 4).
Die Anfallsfreiheitsrate durch VNS wird in systematischen Übersichten mit 74% (28 von 38 Fällen) angegeben, jedoch wurde auf die Möglichkeit eines Publikationsbias hingewiesen4).
Perampanel und Rufinamid haben Wirksamkeit bei nicht-mitochondrialer myoklonischer Epilepsie gezeigt, und es wird erwartet, dass sie bei MERRF eingesetzt werden können5).
Als weitere alternative Therapien werden Atkins-Diät, Glukokortikoide, Cannabidiol (CBD), N-Acetylcystein, tiefe Hirnstimulation und transkranielle Magnetstimulation (30–40% Reduktion der Anfallshäufigkeit) untersucht5).
Kawazoe et al. (2022) berichteten über eine 68-jährige Japanerin mit einer homoplasmischen Mutation m.5820C>A im MT-TC-Gen2). Diese Mutation befindet sich an der Basis des Aminoacyl-Akzeptorstamms der tRNA-Cystein und wurde durch In-silico-Analysen als pathogen vorhergesagt. In diesem Fall zeigte eine Hautbiopsie p62-positive nukleäre Einschlusskörperchen, was einen neuen Zusammenhang zwischen mitochondrialen Erkrankungen und nukleären Einschlusskörperchen aufzeigt2).
QWas ist eine Vagusnervstimulationstherapie?
A
Bei dieser Behandlungsmethode wird eine Elektrode um den linken Vagusnerv im Halsbereich gewickelt und ein Stimulator unter der Brusthaut implantiert, der periodisch elektrische Impulse sendet. Die Wirkung erfolgt über den Hirnstamm und entfaltet eine antikonvulsive Wirkung. Es gibt einen Fallbericht, der die Wirksamkeit bei superrefraktärem Status epilepticus bei MERRF zeigt4), jedoch ist die Evidenz begrenzt.
Jeeva-Patel T, Freund P, Margolin EA. Lipomatosis and optic neuropathy clinches the diagnosis of myoclonic epilepsy with ragged red fibres (MERRF) syndrome. BMJ Case Rep. 2021;14:e240463.
Kawazoe T, Tobisawa S, Sugaya K, et al. Myoclonic epilepsy with ragged-red fibers with intranuclear inclusions. Intern Med. 2022;61:547-552.
Ripolone M, Zanotti S, Napoli L, et al. MERRF mutation A8344G in a four-generation family without central nervous system involvement: clinical and molecular characterization. J Pers Med. 2023;13:147.
Baysal L, Jobi S, Zimmermann S, Helmers A-K, Margraf NG. Successful application of vagus nerve stimulation in super refractory status epilepticus associated with MERRF syndrome. Epilepsy Behav Rep. 2025;30:100769. doi:10.1016/j.ebr.2025.100769.
Finsterer J. A review of the advances in the medical management of epilepsy associated with myoclonic epilepsy with ragged-red fibers (MERRF) syndrome. Cureus. 2025;17(4):e82875.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.