La epilepsia mioclónica con fibras rojas rasgadas (MERRF) es una enfermedad mitocondrial multisistémica rara causada por mutaciones en el ADN mitocondrial. En la CIE-10 se clasifica como E88.4 (trastorno del metabolismo mitocondrial).
Aproximadamente el 80% de los casos de MERRF son causados por la mutación puntual m.8344A>G en el gen MT-TK5). Esta mutación provoca una disfunción del ARNt de lisina, alterando el ensamblaje de proteínas necesarias para la fosforilación oxidativa1). Otras mutaciones causantes reportadas incluyen m.8356T>C, m.8363G>A y m.3243A>G5). También se ha reportado una nueva mutación en el gen MT-TC (m.5820C>A)2).
En casos hereditarios, la transmisión es casi siempre materna. Las enfermedades representativas de la encefalomiopatía mitocondrial incluyen la oftalmoplejía externa progresiva crónica (CPEO), MELAS y MERRF, siendo MELAS la más frecuente.
La prevalencia de MERRF se estima en menos de 1 por cada 100,000 personas. La edad promedio de inicio es de 45 años5), aunque también hay casos de inicio temprano en la infancia con crisis epilépticas. Se ha reportado que el 92.3% de los portadores de la mutación m.8344A>G desarrollan epilepsia4).
La expresión clínica varía ampliamente según el grado de heteroplasmia (proporción de ADNmt mutante y silvestre en la misma célula)1)3). Cuando la carga mutacional es baja, puede haber formas leves sin síntomas del sistema nervioso central. También se han reportado familias que no presentaron síntomas del sistema nervioso central durante cuatro generaciones3).
Q¿Cómo se hereda el MERRF?
A
El ADN mitocondrial se hereda solo de la madre, por lo que sigue un patrón de herencia materna. La proporción de mtDNA mutante (heteroplasmia) varía entre individuos y tejidos, determinando la gravedad de los síntomas clínicos. Consulte la sección «Causas y factores de riesgo» para más detalles.
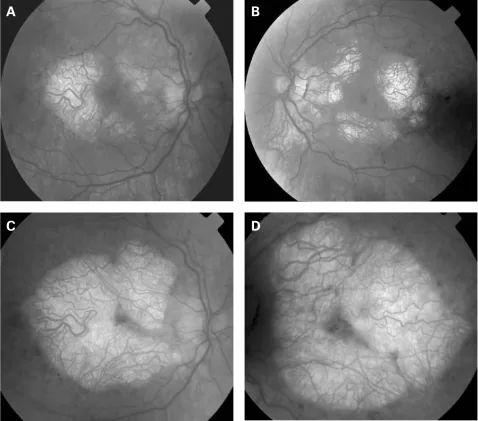
P P Rath, S Jenkins, M Michaelides et al. Characterisation of the macular dystrophy in patients with the A3243G mitochondrial DNA point mutation with fundus autofluorescence. The British Journal of Ophthalmology. 2008 Jan 22; 92(5):623. Figure 2. PMCID: PMC2569141. License: CC BY.
Figura que ilustra los hallazgos oculares en pacientes con MERRF entre 1994 y 2001
Los cuatro signos clásicos del MERRF son mioclonías, epilepsia generalizada, ataxia y fibras rojo-rasgadas en la biopsia muscular. En el ámbito neuroftalmológico, los siguientes síntomas son las principales quejas.
Disminución de la agudeza visual: ocurre de forma progresiva debido a la atrofia óptica. Suele ser bilateral y simétrica.
Mioclonías: contracciones y relajaciones musculares rápidas e involuntarias que generalmente duran de 1 a 2 segundos 5). Pueden ser provocadas por estímulos luminosos 2).
Crisis epilépticas: las crisis mioclónicas son las más frecuentes, pero también se han reportado crisis tónico-clónicas generalizadas, crisis focales y crisis de ausencia5).
Ataxia cerebelosa: la ataxia y la disartria aparecen de forma progresiva4).
Hipoacusia neurosensorial: se presenta con alta frecuencia.
Entumecimiento periférico: trastorno sensorial distal debido a neuropatía periférica2).
Debilidad muscular y fatiga fácil: debido a miopatía4).
Los principales hallazgos neuroftalmológicos asociados con MERRF son cinco: atrofia/neuropatía óptica, oftalmoplejía, ptosis palpebral, retinopatía pigmentaria y nistagmo.
Atrofia óptica
Frecuencia: 39% en pacientes con MERRF (Hirano et al., 13/36 casos), 10% en total de portadores de la mutación m.8344A>G (Altmann et al., 34 casos) 1)
Hallazgos: Palidez bilateral y simétrica del disco óptico1). Un estudio reportó atrofia óptica en 6 de 7 (85.7%) niños y adultos jóvenes confirmados genéticamente.
Punto destacado: Se detectó adelgazamiento de la capa de fibras nerviosas de la retina (RNFL) y de la capa de células ganglionares maculares (GCC) en los 3 pacientes asintomáticos visualmente.
Oftalmoplejía y ptosis palpebral
Oftalmoplejía: 2 de 34 casos (5.9%). La restricción de los movimientos oculares externos varía según los informes: 2/2 casos (Zhu et al.), 3/7 casos (42.9%, Grönlund et al.).
Ptosis palpebral: 10 de 34 casos (29.4%, Mancuso et al.). 1 de 7 casos (Grönlund et al.).
Retinopatía y nistagmo
Retinopatía pigmentaria: 4 de 24 casos (16.7%, Mancuso et al.). Distrofia retiniana en 1 de 7 casos (Grönlund et al.).
Q¿Existe alguna anomalía en el nervio óptico aunque la visión sea normal?
A
Incluso en pacientes visualmente asintomáticos, la OCT puede detectar adelgazamiento de la capa de fibras nerviosas de la retina y del complejo de células ganglionares de la mácula. Esto refleja una pérdida potencial de células retinianas, por lo que se recomienda un seguimiento oftalmológico regular.
Fibras rojo-rasgadas: se confirman mediante biopsia muscular en el 96% de las mutaciones m.8344A>G1).
Lipomatosis múltiple: varía entre 3 y 32.4% según los informes1). Se ha señalado una asociación entre la alta heteroplasmia y el tamaño de los lipomas1).
Miocardiopatía, diabetes mellitus e hipotiroidismo: se presentan como parte de la afectación multiorgánica5).
Acidosis láctica: se observa un aumento del lactato en el líquido cefalorraquídeo2).
Discapacidad intelectual y demencia: aparecen en casos avanzados.
La causa del MERRF es una mutación en el ADN mitocondrial (ADNmt).
Mutación m.8344A>G en el gen MT-TK: es la más frecuente, representando aproximadamente el 80% de todos los casos5). Provoca una anomalía estructural del ARNt de lisina, alterando la síntesis de proteínas necesarias para la fosforilación oxidativa1).
Otras mutaciones del ADNmt: m.8356T>C, m.8363G>A, m.3243A>G, m.3255G>A, m.5820C>A en el gen MT-TC, entre otras2)5).
Herencia materna: dado que el ADNmt se hereda exclusivamente de la madre, en las familias afectadas la transmisión ocurre solo por vía materna.
Heteroplasmia y efecto umbral: cuando la proporción de mtDNA mutante supera el umbral específico del tejido, aparecen anomalías bioquímicas 3). Se estima que el umbral en el músculo esquelético es del 60-90% de mutación, y si queda aproximadamente un 15% de mtDNA de tipo salvaje, la traducción y la actividad de la COX se recuperan casi por completo 3).
Genes modificadores nucleares: se sugiere la existencia de factores que afectan la segregación y replicación del mtDNA, regulando la distribución de la cantidad de mutación entre los tejidos 3).
Las pruebas genéticas moleculares son esenciales para el diagnóstico definitivo. En más del 80% de los pacientes se identifica la mutación m.8344A>G 5).
Muestra: Extracción de ADN de sangre, orina o músculo
Método: Cuantificación de heteroplasmia mediante PCR-RFLP 3)
Nota: Generalmente, la heteroplasmia en orina es mayor que en sangre 3)
Con la modificación de Gomori de tricrómico se confirman fibras rojas rasgadas. Se observan fibras con tinción intensa de SDH y deficiencia de COX. En la doble tinción COX-SDH, aproximadamente el 5% de las fibras muestran deficiencia de COX 2). En microscopía electrónica se observa proliferación y elongación anormal de mitocondrias debajo del sarcolema 2).
Estudios de conducción nerviosa: muestran un patrón de neuropatía sensorial 2)
Electroencefalograma (EEG): se observa fotosensibilidad. Los potenciales evocados somatosensoriales gigantes (SEP 90 μV, normal <10 μV) son característicos 2)
Electromiografía: cambios miogénicos en extremidades inferiores distales 2)
Tomografía de coherencia óptica (OCT): puede detectar adelgazamiento de la capa de fibras nerviosas de la retina y del complejo de células ganglionares de la retina incluso en pacientes asintomáticos1)
Examen de fondo de ojo: palidez del disco óptico1)
En las etapas iniciales, a menudo se diagnostica erróneamente como JME, y los síntomas sistémicos acompañantes como la ataxia y el deterioro cognitivo progresivo son útiles para el diagnóstico diferencial de PME1).
Actualmente no existe un tratamiento curativo para MERRF. El tratamiento se centra en la terapia sintomática, y un enfoque multidisciplinario que combine el manejo de la epilepsia con cuidados de apoyo sistémicos es fundamental.
Para la epilepsia mioclónica, el levetiracetam se considera el más eficaz5).
Terapia combinada de levetiracetam y clonazepam: en un estudio de 17 pacientes con MERRF, los 12 que cambiaron de monoterapia a terapia combinada mostraron mejoría. También se ha informado mejoría en la función cognitiva y la coordinación motora5).
Clonazepam: como benzodiazepina, es eficaz por sí solo para la mioclonía5).
Zonisamida: se ha sugerido su eficacia5).
Piracetam: eficaz en algunos casos5). En el caso de una mujer japonesa de 68 años, tras suspender fenitoína y carbamazepina, se cambió a un régimen basado en piracetam y levetiracetam, mejorando los síntomas2).
FAE
Efecto sobre la mioclonía
Precauciones
Levetiracetam
Eficaz
Primera línea
Clonazepam
Eficaz
Sedación
Zonisamida
Posible eficacia
—
Piracetam
Posible eficacia
—
Ácido valproico
—
Contraindicado en principio (toxicidad mitocondrial)
Es necesario un seguimiento regular por parte de neurología, oftalmología, cardiología y endocrinología 1). Se recomienda evaluación clínica, análisis de sangre, EEG y monitorización de niveles en sangre de ASM cada tres meses 5).
Q¿Por qué no se puede usar valproato en MERRF?
A
Se ha confirmado experimentalmente que el valproato reduce la actividad de los complejos I y IV de la cadena respiratoria mitocondrial, y en enfermedades mitocondriales como MERRF existe el riesgo de empeorar la patología 5). En el tratamiento de la epilepsia, se prefieren fármacos con baja toxicidad mitocondrial, como levetiracetam o clonazepam.
6. Fisiopatología y mecanismo detallado de la enfermedad
La mutación m.8344A>G en el gen MT-TK altera la estructura del ARNt de lisina, lo que afecta la síntesis de proteínas dentro de la mitocondria (ensamblaje de los complejos de fosforilación oxidativa) 1). Como resultado, disminuye la producción de ATP y los órganos con alta demanda energética (cerebro, músculo esquelético, miocardio) se ven afectados de manera preferente 3).
Las mutaciones del ADN mitocondrial existen en estado de heteroplasmia, y cuando la proporción de mtDNA mutante supera un umbral específico del tejido, aparece el fenotipo bioquímico 3).
Umbral patológico del músculo esquelético: 60-90% de mutación 3)
Con aproximadamente un 15% de mtDNA silvestre residual, la traducción y la actividad de la COX pueden recuperarse a niveles casi normales 3)
Los genes modificadores nucleares regulan la segregación y replicación del mtDNA de manera específica del tejido 3)
Como reacción compensatoria a la deficiencia de COX (disfunción del complejo IV de la cadena respiratoria), las mitocondrias anormales proliferan bajo el sarcolema, formando fibras rojo rasgadas (RRF) 2). En la microscopía electrónica se observa una marcada proliferación de mitocondrias anormalmente alargadas y cuerpos de inclusión paracristalinos 2).
Las células ganglionares de la retina tienen una alta demanda energética y son vulnerables a la disfunción mitocondrial 1). Incluso en pacientes visualmente asintomáticos, se detecta adelgazamiento de la RNFL y la GCC, lo que sugiere que está ocurriendo una pérdida neuronal subyacente.
La disfunción mitocondrial provoca un aumento del estrés oxidativo, anomalías del sistema inmunitario y alteraciones en la dinámica mitocondrial, y también está implicada en la patogénesis de las crisis epilépticas4).
7. Investigación reciente y perspectivas futuras (informes en fase de investigación)
Baysal et al. (2025) implantaron un dispositivo de VNS en una mujer de 38 años con la mutación m.8344A>G que presentaba estado epiléptico superrefractario (SRSE) y aplicaron un método de titulación rápida (aumento hasta 2 mA en 6 días)4). Siete días después de la implantación del VNS, comenzó la mejoría de las mioclonías, y a los 21 días desaparecieron las crisis tónico-clónicas generalizadas. Durante un seguimiento de dos años, no hubo recurrencia de crisis tónico-clónicas generalizadas ni estado epiléptico, y las mioclonías también disminuyeron. Se estima que el mecanismo de acción del VNS implica la activación de la vía núcleo del tracto solitario → locus coeruleus (liberación de norepinefrina) y núcleos del rafe (liberación de serotonina) a través de la estimulación del nervio vago izquierdo, así como la regulación al alza de la densidad de receptores GABA-A4).
La tasa de cese del SE con VNS se ha reportado en una revisión sistemática como del 74% (28 de 38 casos), aunque se ha señalado la posibilidad de sesgo de publicación4).
Se ha informado de la eficacia de perampanel y rufinamida en la mioclonía epiléptica no mitocondrial, y se espera su aplicación en MERRF5).
Como otras terapias alternativas, se están considerando la dieta Atkins, glucocorticoides, cannabidiol (CBD), N-acetilcisteína, estimulación cerebral profunda y estimulación magnética transcraneal (reducción del 30-40% en la frecuencia de las convulsiones)5).
Kawazoe et al. (2022) reportaron el caso de una mujer japonesa de 68 años con la mutación homoplásmica m.5820C>A en el gen MT-TC2). Esta mutación se localiza en la base del brazo aceptor de aminoácidos del ARNt de cisteína, y se predijo su patogenicidad mediante análisis in silico. En este caso, se observaron inclusiones intranucleares positivas para p62 en una biopsia de piel, lo que sugiere una nueva asociación entre enfermedades mitocondriales y las inclusiones intranucleares2).
Q¿Qué es la terapia de estimulación del nervio vago?
A
Es un tratamiento que consiste en enrollar un electrodo alrededor del nervio vago en el lado izquierdo del cuello y enviar estimulación eléctrica periódica desde un dispositivo implantado debajo de la piel del pecho. Actúa a través del tronco encefálico para ejercer un efecto anticonvulsivo. Existe un informe de caso en el que fue eficaz para el estado epiléptico superrefractario en MERRF4), pero la evidencia es limitada.
Jeeva-Patel T, Freund P, Margolin EA. Lipomatosis and optic neuropathy clinches the diagnosis of myoclonic epilepsy with ragged red fibres (MERRF) syndrome. BMJ Case Rep. 2021;14:e240463.
Kawazoe T, Tobisawa S, Sugaya K, et al. Myoclonic epilepsy with ragged-red fibers with intranuclear inclusions. Intern Med. 2022;61:547-552.
Ripolone M, Zanotti S, Napoli L, et al. MERRF mutation A8344G in a four-generation family without central nervous system involvement: clinical and molecular characterization. J Pers Med. 2023;13:147.
Baysal L, Jobi S, Zimmermann S, Helmers A-K, Margraf NG. Successful application of vagus nerve stimulation in super refractory status epilepticus associated with MERRF syndrome. Epilepsy Behav Rep. 2025;30:100769. doi:10.1016/j.ebr.2025.100769.
Finsterer J. A review of the advances in the medical management of epilepsy associated with myoclonic epilepsy with ragged-red fibers (MERRF) syndrome. Cureus. 2025;17(4):e82875.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.