视神经萎缩
频率:MERRF患者中39%(Hirano等13/36例),m.8344A>G突变携带者总体中10%(Altmann等34例)1)
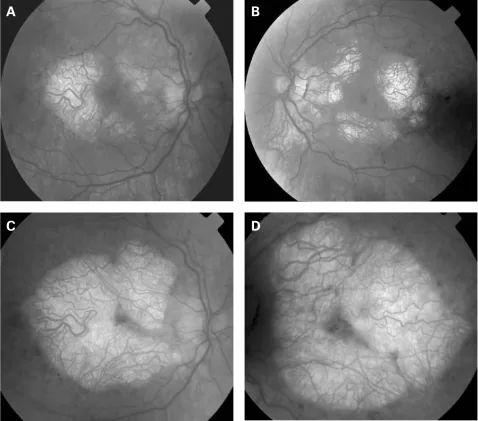
表现:双侧对称性视盘苍白1)。有报告称遗传学确诊的儿童及年轻成人7例中6例(85.7%)存在视神经萎缩。
注意点:所有3例视觉无症状患者均检测到RNFL和黄斑GCC变薄。

伴有破碎红纤维的肌阵挛癫痫(MERRF)是一种由线粒体DNA突变引起的罕见多系统线粒体病。ICD-10分类为E88.4(线粒体代谢障碍)。
MERRF约80%由MT-TK基因的m.8344A>G点突变引起5)。该突变导致tRNA赖氨酸功能异常,阻碍氧化磷酸化所需蛋白质的组装1)。其他致病突变包括m.8356T>C、m.8363G>A、m.3243A>G等5)。MT-TC基因的新突变(m.5820C>A)也有报道2)。
遗传方式几乎均为母系遗传。线粒体脑肌病的代表性疾病包括慢性进行性眼外肌麻痹(CPEO)、MELAS和MERRF,其中MELAS最为常见。
MERRF的患病率估计低于十万分之一。平均发病年龄为45岁5),但也有在婴幼儿早期以癫痫发作发病的病例。据报道,m.8344A>G突变携带者中92.3%会出现癫痫4)。
异质性(同一细胞内突变型与野生型mtDNA的混合比例)程度不同,临床表现差异很大1)3)。突变负荷低时,可能出现无中枢神经系统症状的轻型。也有报告称,一个家系连续四代未出现中枢神经系统临床症状3)。
线粒体DNA仅由母亲遗传,因此呈母系遗传模式。突变型mtDNA的比例(异质性)因个体和组织而异,影响临床症状的严重程度。详情请参阅“原因与风险因素”章节。
MERRF的经典四联征包括肌阵挛、全身性癫痫、共济失调以及肌肉活检中的破碎红纤维。在神经眼科领域,以下症状为主诉。
MERRF相关的主要神经眼科所见包括视神经萎缩/视神经病变、眼肌麻痹、眼睑下垂、色素性视网膜病变和眼球震颤五种。
视神经萎缩
频率:MERRF患者中39%(Hirano等13/36例),m.8344A>G突变携带者总体中10%(Altmann等34例)1)
表现:双侧对称性视盘苍白1)。有报告称遗传学确诊的儿童及年轻成人7例中6例(85.7%)存在视神经萎缩。
注意点:所有3例视觉无症状患者均检测到RNFL和黄斑GCC变薄。
眼肌麻痹·眼睑下垂
眼肌麻痹:34例中2例(5.9%)。外眼肌运动受限的报告存在差异:2/2例(Zhu等),7例中3例(42.9%,Grönlund等)。
眼睑下垂:34例中10例(29.4%,Mancuso等)。7例中1例(Grönlund等)。
视网膜病变·眼球震颤
色素性视网膜病变:24例中4例(16.7%,Mancuso等)。7例中1例视网膜营养不良(Grönlund等)。
眼震:7例中有1例(Grönlund等)。
即使视觉上无症状的患者,OCT检查也可能检测到视网膜神经纤维层和黄斑区神经节细胞复合体变薄。这反映了潜在的视网膜神经细胞丢失,建议定期眼科随访。
MERRF的病因是线粒体DNA(mtDNA)突变。
MERRF的诊断采用以下4个临床特征(经典四联征)。
确诊需要进行分子遗传学检测。超过80%的患者可检测到m.8344A>G突变5)。
Gomori三色改良染色可见破碎红纤维。可见SDH强染色、COX缺失纤维。COX-SDH双重染色中约5%可见COX缺失纤维2)。电子显微镜下可见肌膜下线粒体异常增殖和伸长2)。
乳酸19 mg/dL(正常4.2~17.0)、丙酮酸1.2 mg/dL(正常0.3~0.9)轻度升高2)。
骨骼肌的复合体IV活性降至对照的55%~69%2)。
MERRF是进行性肌阵挛癫痫(PME)的一种类型,需与以下疾病进行鉴别。
| 疾病 | 鉴别要点 |
|---|---|
| 青少年肌阵挛癫痫(JME) | 无认知功能下降或共济失调进展,预后良好 |
| Lafora病 | 皮肤活检可见Lafora小体阳性 |
| Unverricht-Lundborg病 | CSTB基因突变,进展缓慢 |
| 神经元蜡样脂褐质沉积症 | 视网膜变性显著,酶缺乏 |
早期易误诊为JME,共济失调和认知功能下降等全身伴随症状有助于PME的鉴别1)。
目前MERRF尚无根治方法。治疗以对症治疗为主,基本采用癫痫管理与全身支持治疗相结合的多学科方法。
对于肌阵挛癫痫,左乙拉西坦被认为最有效5)。
| ASM | 对肌阵挛的效果 | 注意事项 |
|---|---|---|
| 左乙拉西坦 | 有效 | 一线用药 |
| 氯硝西泮 | 有效 | 镇静作用 |
| 唑尼沙胺 | 可能有效 | — |
| 吡拉西坦 | 可能有效 | — |
| 丙戊酸 | — | 原则上禁忌(线粒体毒性) |
| 苯妥英 | 肌阵挛恶化 | 避免使用 |
| 卡马西平 | 肌阵挛恶化 | 避免使用 |
为辅助线粒体功能,经验性推荐以下补充剂,但缺乏强有力的证据1)。
推荐进行物理治疗和有氧运动以维持运动能力。作业治疗和言语治疗也有帮助4)。
需要神经内科、眼科、心内科和内分泌科的定期随访1)。建议每3个月进行临床评估、血液检查、脑电图和ASM血药浓度监测5)。
实验证实丙戊酸会降低线粒体呼吸链复合物I和IV的活性,在MERRF等线粒体疾病中可能加重病情5)。癫痫治疗应优先选用左乙拉西坦、氯硝西泮等线粒体毒性较低的药物。
MERRF的病理源于mtDNA突变导致的线粒体功能障碍。
MT-TK基因的m.8344A>G突变改变了tRNA赖氨酸的结构,损害线粒体内的蛋白质合成(氧化磷酸化复合物的组装)1)。结果导致ATP生成减少,能量需求高的器官(脑、骨骼肌、心肌)优先受损3)。
线粒体DNA突变以异质状态存在,当突变型mtDNA的比例超过组织特异性阈值时,会出现生化表型3)。
作为对COX缺乏(呼吸链复合体IV障碍)的代偿反应,肌膜下异常线粒体增殖,形成破碎红纤维(RRF)2)。电子显微镜下可见异常延长的线粒体显著增殖和副结晶体包涵体2)。
视网膜神经节细胞能量需求高,对线粒体功能障碍脆弱1)。即使在视觉无症状的患者中,也能检测到RNFL和GCC变薄,提示潜在的神经细胞丢失正在进展。
线粒体功能障碍会导致氧化应激增加、免疫系统异常和线粒体动力学改变,并参与癫痫发作的病理过程4)。
Baysal等人(2025)对一名携带m.8344A>G突变的38岁女性超难治性癫痫持续状态(SRSE)患者植入了VNS装置并采用快速递增法(6天内增加至2 mA)4)。VNS植入7天后肌阵挛开始改善,21天后全面性强直-阵挛发作消失。2年随访期间无全面性强直-阵挛发作或癫痫持续状态复发,肌阵挛也减少。VNS的作用机制推测为通过刺激左迷走神经激活孤束核→蓝斑核(释放去甲肾上腺素)和中缝核(释放5-羟色胺)通路,并上调GABA-A受体密度4)。
VNS终止SE的比率在系统综述中报告为74%(38例中28例),但存在报告偏倚的可能性4)。
据报道,吡仑帕奈和卢非酰胺在非线粒体性肌阵挛癫痫中有效,有望应用于MERRF5)。
其他替代疗法包括阿特金斯饮食、糖皮质激素、大麻二酚(CBD)、N-乙酰半胱氨酸、脑深部刺激、经颅磁刺激(可降低30-40%的发作频率)正在研究中5)。
Kawazoe等人(2022)报告了一名68岁日本女性,携带MT-TC基因的同质突变m.5820C>A2)。该突变位于tRNA半胱氨酸的氨基酸受体茎的根部,计算机模拟分析预测其具有致病性。该病例的皮肤活检发现p62阳性核内包涵体,提示了线粒体疾病与核内包涵体之间的新关联2)。
这是一种将电极缠绕在左颈部迷走神经上,通过植入胸部皮下的刺激装置定期发送电刺激的治疗方法。它通过脑干发挥抗惊厥作用。有病例报告显示其对MERRF的超难治性癫痫持续状态有效4),但证据有限。