Outras Cataratas Congênitas
Morfologia: Polar anterior, polar posterior, lamelar, catarata total, etc.
Ponto de diferenciação: Localização, forma e cor da opacidade são diferentes.
Exame: Exame com lâmpada de fenda, histórico familiar.

A catarata cerúlea (cerulean cataract) é uma catarata de desenvolvimento hereditária caracterizada por opacidades branco-azuladas dispersas no núcleo e córtex do cristalino. Também chamada de “blue dot cataract”. As opacidades nucleares são classificadas por forma em pulverulentas, medusoides, dendríticas, etc., mas a catarata cerúlea é uma classificação baseada na cor da opacidade.
Segue um padrão de herança autossômica dominante. A progressão é lenta e muitas vezes não afeta a visão até a idade adulta. No entanto, em alguns pacientes, ocorre deficiência visual precoce e, se não tratada, pode levar à ambliopia ou nistagmo.
Sabe-se que está associada à síndrome de Down. Crianças com síndrome de Down podem desenvolver catarata congênita ou adquirida, e particularmente a catarata cerúlea (composta por depósitos de amiloide) foi relatada em associação1).
Crianças com síndrome de Down podem desenvolver catarata congênita ou adquirida. Particularmente, a catarata cerúlea composta por depósitos de amiloide foi relatada em associação1). A síndrome de Down também é conhecida por estar associada ao ceratocone (até 15% dos adultos), e casos de ectasia corneana e catarata cerúlea coexistindo no mesmo paciente foram relatados1).
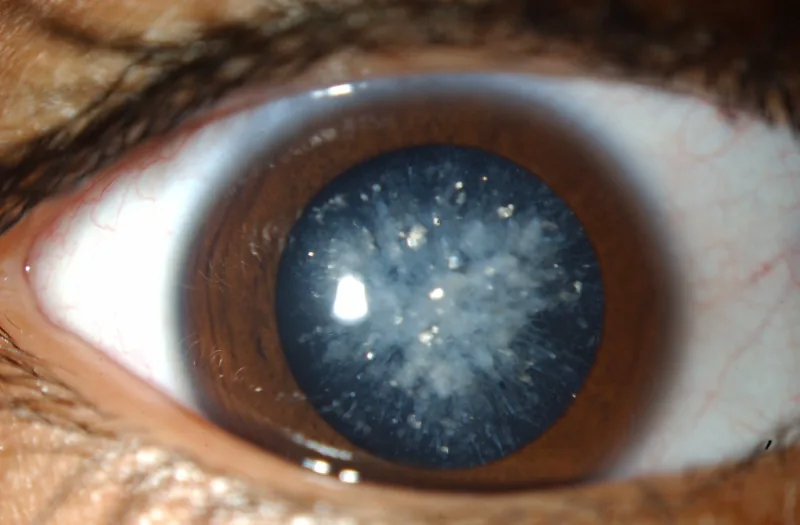
O exame com lâmpada de fenda revela pequenas opacidades azuis ou brancas na camada superficial do núcleo do cristalino. As opacidades começam no núcleo embrionário e se espalham pelo núcleo e córtex. Geralmente são bilaterais.
Em casos associados à síndrome de Down, podem ser encontrados simultaneamente sinais de ceratocone, como anel de Fleischer e protrusão cônica da córnea 1). Casos com edema corneano agudo também foram relatados 1).
Na classificação morfológica da catarata congênita, incluem-se os tipos: capsular, polar, nuclear, lamelar, punctata, sutural e total. A catarata azul é classificada como um subtipo de catarata nuclear.
A catarata azul segue um padrão de herança autossômica dominante. Pelo menos quatro loci gênicos foram identificados.
| Locus Gênico | Localização Cromossômica |
|---|---|
| CCA1 | 17q24 |
| CCA2 | 22q11.2-q12.2 |
| CCA3 | 2q33-q35 |
| CC4 | 16q22-q23 |
Mutações nos genes da cristalina β-B2 (CRYBB2), cristalina γ-D (CRYGD), gene homólogo do oncogene do fibrossarcoma aponeurótico muscular aviário V-MAF (MAF) e proteína intrínseca principal das fibras do cristalino (MIP) foram identificadas como genes causadores.
Quanto à etiologia geral da catarata congênita, a mais comum é idiopática (30-50%), seguida por hereditária (autossômica dominante é a mais comum), infecção intrauterina, distúrbios metabólicos, anomalias cromossômicas, doenças oculares associadas e doenças ou síndromes sistêmicas associadas.
Mutações em CRYBB2 (β-B2-cristalina), CRYGD (γ-D-cristalina), MAF e MIP (proteína intrínseca principal das fibras do cristalino) foram identificadas. Esses genes codificam proteínas estruturais do cristalino e fatores de transcrição, e as mutações prejudicam a transparência do cristalino. Os loci gênicos conhecidos são quatro regiões: 17q24, 22q11.2-q12.2, 2q33-q35, 16q22-q23.
Este é o exame básico para o diagnóstico. Observam-se pequenas opacidades azuis ou brancas na superfície do núcleo do cristalino. São detectadas ao nascimento ou durante exames oftalmológicos de rotina.
Outras Cataratas Congênitas
Morfologia: Polar anterior, polar posterior, lamelar, catarata total, etc.
Ponto de diferenciação: Localização, forma e cor da opacidade são diferentes.
Exame: Exame com lâmpada de fenda, histórico familiar.
Catarata de Desenvolvimento
Morfologia: Opacidade que progride após o nascimento.
Ponto de diferenciação: Velocidade de progressão e presença de complicações sistêmicas.
Exame: Exame seriado com lâmpada de fenda ao longo do tempo.
Catarata Traumática
Morfologia: Opacidade secundária a trauma.
Ponto de diferenciação: Presença de histórico de trauma.
Exame: Anamnese, exame do segmento anterior.
A história familiar é importante para o diagnóstico diferencial. O teste genético pode identificar a mutação causadora em alguns casos.
Atualmente não existe tratamento para prevenir a formação ou progressão da catarata azul. Avaliações oftalmológicas regulares são realizadas para monitorar a progressão da deficiência visual.
Quando a diminuição da visão progride e interfere nas atividades diárias, a cirurgia de catarata (facoemulsificação) e o implante de lente intraocular são indicados. A função visual após o implante de lente intraocular é boa, e a cirurgia é realizada ativamente se a melhora visual for considerada certa.
Na catarata congênita unilateral com deficiência visual grave, a cirurgia precoce é necessária. A intervenção no momento adequado é importante do ponto de vista da prevenção da ambliopia. Em casos com complicações oculares graves ou doenças do sistema nervoso central, a melhora da função visual não é esperada, portanto a indicação cirúrgica é cuidadosamente avaliada.
A cirurgia de catarata é indicada quando a diminuição da visão progride e interfere nas atividades diárias ou no desenvolvimento visual. Na maioria dos pacientes, não afeta a visão até a idade adulta, portanto é manejada com observação regular até lá. Em crianças com deficiência visual precoce, a intervenção cirúrgica no momento adequado é importante para prevenir ambliopia.
A catarata azul é causada por mutações nos genes das cristalinas. As cristalinas são as principais proteínas estruturais do cristalino, essenciais para manter a transparência e a função refrativa do cristalino.
Mutações em CRYBB2 e CRYGD prejudicam o dobramento normal das proteínas do cristalino. A agregação de proteínas anormais leva à perda de transparência do cristalino, causando uma opacidade azul-esbranquiçada característica. O gene MAF codifica um fator de transcrição envolvido no desenvolvimento e diferenciação do cristalino. O gene MIP codifica a principal proteína de membrana das fibras do cristalino, contribuindo para a homeostase do cristalino.
Na catarata azul associada à síndrome de Down, foi relatado que a opacidade consiste em depósitos de amiloide 1). A síndrome de Down também é conhecida por estar associada ao ceratocone, e foi sugerido que o hipotireoidismo concomitante pode contribuir para a progressão da ectasia corneana 1).