Altre cataratte congenite
Forma: polare anteriore, polare posteriore, lamellare, cataratta completa, ecc.
Punto di differenziazione: sede, forma e colore dell’opacità sono diversi.
Esame: esame con lampada a fessura, anamnesi familiare.

La cataratta cerulea (cerulean cataract) è una cataratta dello sviluppo ereditaria caratterizzata da opacità bianco-azzurre disseminate nel nucleo e nella corteccia del cristallino. È anche chiamata ‘cataratta a punti blu’. Le opacità nucleari sono classificate in base alla forma in polverulenta, medusoide, dendritica, ecc., ma la cataratta cerulea è una classificazione basata sul colore delle opacità.
Segue un modello di ereditarietà autosomica dominante. La progressione è lenta e spesso non influisce sulla vista fino all’età adulta. Tuttavia, in alcuni pazienti si verifica un deficit visivo precoce e, se non trattato, può portare ad ambliopia e nistagmo.
È nota un’associazione con la sindrome di Down. I bambini con sindrome di Down possono sviluppare cataratta congenita o acquisita, in particolare è stata riportata la presenza di cataratta cerulea (costituita da depositi di amiloide) 1).
I bambini con sindrome di Down possono sviluppare cataratta congenita o acquisita. In particolare, è stata riportata la presenza di cataratta cerulea dovuta a depositi di amiloide 1). La sindrome di Down è anche associata al cheratocono (fino al 15% degli adulti) e sono stati riportati casi di coesistenza di ectasia corneale e cataratta cerulea nello stesso paziente 1).
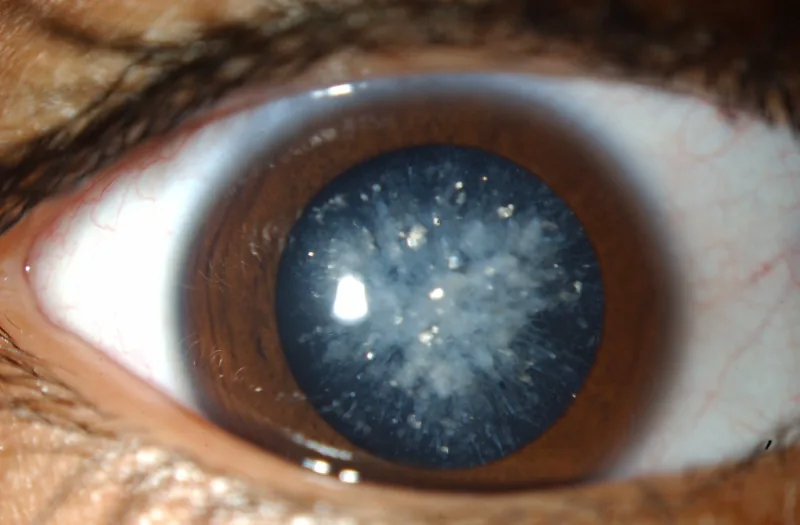
L’esame con lampada a fessura rivela minuscole opacità blu o bianche nello strato superficiale del nucleo del cristallino. Le opacità iniziano nel nucleo embrionale e sono sparse nel nucleo e nella corteccia. Di solito sono bilaterali.
Nei pazienti con sindrome di Down, si possono osservare contemporaneamente segni di cheratocono come l’anello di Fleischer e la protrusione conica della cornea1). Sono stati riportati anche casi complicati da edema corneale acuto1).
Nella classificazione morfologica delle cataratte congenite si distinguono cataratta capsulare, polare, nucleare, lamellare, puntata, suturale e totale. La cataratta blu è classificata come un sottotipo della cataratta nucleare.
La cataratta blu segue un modello di ereditarietà autosomica dominante. Sono stati identificati almeno quattro loci genetici.
| Locus genetico | Posizione cromosomica |
|---|---|
| CCA1 | 17q24 |
| CCA2 | 22q11.2-q12.2 |
| CCA3 | 2q33-q35 |
| CC4 | 16q22-q23 |
Sono state identificate mutazioni nei geni della β-B2-cristallina (CRYBB2), della γ-D-cristallina (CRYGD), del gene omologo dell’oncogene del fibrosarcoma muscolo-aponeurotico aviario V-MAF (MAF) e del gene della proteina intrinseca maggiore delle fibre del cristallino (MIP).
Per quanto riguarda l’eziologia complessiva della cataratta congenita, la più frequente è idiopatica (30-50%), seguita da cause ereditarie (autosomica dominante la più comune), infezioni intrauterine, anomalie metaboliche, anomalie cromosomiche, concomitanza di malattie oculari e concomitanza di malattie/sindromi sistemiche.
Sono state identificate mutazioni in CRYBB2 (β-B2-cristallina), CRYGD (γ-D-cristallina), MAF e MIP (proteina intrinseca maggiore delle fibre del cristallino). Questi geni codificano per proteine strutturali del cristallino o fattori di trascrizione; le mutazioni compromettono la trasparenza del cristallino. Sono noti quattro loci genetici: 17q24, 22q11.2-q12.2, 2q33-q35 e 16q22-q23.
Questo è l’esame di base per la diagnosi. Sulla superficie del nucleo del cristallino si osservano piccole opacità blu o bianche. Vengono scoperte alla nascita o durante un esame oculistico di routine.
Altre cataratte congenite
Forma: polare anteriore, polare posteriore, lamellare, cataratta completa, ecc.
Punto di differenziazione: sede, forma e colore dell’opacità sono diversi.
Esame: esame con lampada a fessura, anamnesi familiare.
Cataratta dello sviluppo
Forma: opacità che progredisce dopo la nascita.
Punto di differenziazione: velocità di progressione e presenza di complicanze sistemiche.
Esame: esame con lampada a fessura nel tempo.
Cataratta traumatica
Forma: opacità secondaria a trauma.
Punto di differenziazione: presenza o assenza di storia di trauma.
Esame: anamnesi, esame del segmento anteriore.
L’anamnesi familiare è importante per la diagnosi differenziale. Il test genetico può talvolta identificare la mutazione causale.
Attualmente non esiste un trattamento per prevenire la formazione o la progressione della cataratta blu. Vengono effettuate valutazioni oftalmologiche regolari per monitorare la progressione del deficit visivo.
Quando la riduzione dell’acuità visiva progredisce e interferisce con le attività quotidiane, è indicata la chirurgia della cataratta (facoemulsificazione) con impianto di lente intraoculare. La funzione visiva dopo l’impianto di lente intraoculare è buona e la chirurgia viene eseguita attivamente se si prevede un miglioramento certo della funzione visiva.
In caso di cataratta congenita unilaterale con grave deficit visivo, è necessario un intervento chirurgico precoce. Dal punto di vista della prevenzione dell’ambliopia, è importante un intervento tempestivo. Nei casi con gravi complicanze oculari o malattie del sistema nervoso centrale, non è previsto un miglioramento della funzione visiva, quindi l’indicazione chirurgica viene valutata con cautela.
La chirurgia della cataratta è indicata quando la riduzione dell’acuità visiva progredisce e interferisce con le attività quotidiane o lo sviluppo visivo. In molti pazienti la vista non è compromessa fino all’età adulta, quindi fino ad allora viene gestita con regolare follow-up. Nei bambini con deficit visivo precoce, un intervento chirurgico tempestivo è importante per prevenire l’ambliopia.
La cataratta blu è causata da mutazioni nei geni delle cristalline. Le cristalline sono le principali proteine strutturali del cristallino e sono essenziali per mantenere la trasparenza e la funzione di rifrazione del cristallino.
Le mutazioni in CRYBB2 o CRYGD compromettono il normale ripiegamento delle proteine del cristallino. L’aggregazione di proteine anomale porta alla perdita di trasparenza del cristallino e a un caratteristico offuscamento blu-bianco. Il gene MAF codifica un fattore di trascrizione coinvolto nello sviluppo e nella differenziazione del cristallino. Il gene MIP codifica la principale proteina di membrana delle fibre del cristallino e contribuisce al mantenimento dell’omeostasi del cristallino.
Nella cataratta blu associata alla sindrome di Down, è stato riportato che l’opacità è costituita da depositi di amiloide 1). La sindrome di Down è anche nota per essere associata al cheratocono, ed è stato suggerito che l’ipotiroidismo concomitante possa contribuire alla progressione dell’ectasia corneale 1).