La degeneración corneal harinosa (cornea farinata) es un hallazgo en el que aparecen opacidades finas similares a polvo justo por delante de la membrana de Descemet en la capa más profunda del estroma corneal1). Fue descrita por primera vez por el oftalmólogo suizo Arthur Vogt y también se llama “córnea harinosa”.
Ocurre bilateralmente y progresa lentamente con la edad. Como no afecta la visión, su importancia clínica es limitada. Se clasifica como una degeneración, no una distrofia corneal. Los informes en menores de 40 años son raros. La microscopía confocal in vivo (IVCM) muestra partículas altamente reflectantes dentro del citoplasma de los queratocitos en el estroma corneal profundo 1).
Opacidades corneales profundas similares también se observan en pacientes con ictiosis ligada al cromosoma X (XLI) causada por mutaciones en el gen STS (Xp22.31) 2,3). Se encuentran en el 50% de los pacientes y el 25% de las mujeres portadoras, apareciendo desde la edad adulta temprana 2). En este caso, no se debe al envejecimiento sino a la deficiencia de esteroide sulfatasa y la acumulación de sulfato de colesterol 3).
La degeneración corneal harinosa generalmente no requiere tratamiento. Dado que es asintomática y no afecta la visión, solo se necesita observación. Sin embargo, es importante diferenciarla de afecciones como la distrofia endotelial corneal de Fuchs, que también presenta opacidades corneales profundas. La de Fuchs se acompaña de una disminución en la densidad de células endoteliales corneales, mientras que en la degeneración corneal harinosa el endotelio corneal es normal.
Joobin Khadamy Ocular Manifestations Leading to the Diagnosis of Ichthyosis: A Case Report 2025 Mar 4 Cureus.; 17(3):e80023 Figure 2. PMCID: PMC11968076. License: CC BY.
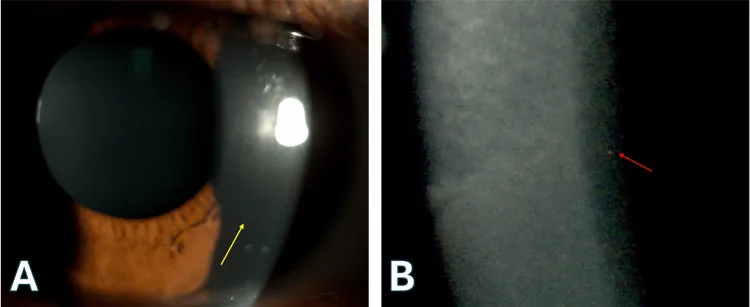
La imagen A es una fotografía del ojo tomada con microscopía de lámpara de hendidura, que muestra nervios corneales elevados (flechas amarillas). La imagen B es una vista ampliada de la misma microscopía de lámpara de hendidura, que muestra opacidades puntiformes en el estroma corneal profundo (flechas rojas). Estos hallazgos son consistentes con un diagnóstico de córnea harinosa (Cornea Farinata).
La degeneración corneal harinosa suele ser asintomática. No causa disminución de la visión, dolor ocular, sensación de cuerpo extraño ni fotofobia. En muchos casos, se descubre incidentalmente durante un examen con lámpara de hendidura.
En la microscopía con lámpara de hendidura, mediante retroiluminación o reflexión especular, se observan depósitos granulares finos de color gris blanquecino a marrón amarillento en la superficie posterior de la córnea. Las opacidades se distribuyen densamente en la córnea central y paracentral, y disminuyen hacia la periferia. Los depósitos individuales son extremadamente pequeños y pueden pasarse por alto fácilmente con la iluminación directa de la hendidura.
Las opacidades se distribuyen difusa y uniformemente en el estroma corneal anterior a la membrana de Descemet. La membrana de Descemet, el epitelio corneal y la capa de células endoteliales son normales. El grosor corneal está dentro del rango normal. La microscopía especular muestra morfología y densidad normales de las células endoteliales corneales.
El factor de riesgo más importante es la edad. Se observa con frecuencia en personas mayores y progresa lentamente, pero rara vez se vuelve clínicamente significativo. Los informes en menores de 40 años son raros.
Mutación del gen STS y ictiosis ligada al cromosoma X
La deficiencia de esteroide sulfatasa debida a mutaciones en el gen STS (Xp22.31) causa ictiosis ligada al cromosoma X (XLI). En pacientes con XLI, el sulfato de colesterol se acumula en el estroma corneal profundo, produciendo opacidades similares a la degeneración corneal farinata. Se han reportado al menos seis mutaciones diferentes en el gen STS, y el tipo de mutación afecta la expresión y función de la enzima esteroide sulfatasa, dando lugar a diversos fenotipos.
Retroiluminación: El método más básico para visualizar depósitos granulares finos en la superficie posterior de la córnea.
Reflejo especular: Permite observar con alto contraste las opacidades justo anteriores a la membrana de Descemet.
Iluminación directa: Los depósitos pequeños se pasan por alto fácilmente, por lo que es importante combinarla con la retroiluminación.
Microscopía especular
Evaluación del endotelio corneal: Confirma que la morfología y densidad de las células endoteliales son normales.
Diferenciación de la distrofia de Fuchs: En la distrofia de Fuchs se observan gutas y disminución de la densidad celular endotelial, mientras que en esta enfermedad el endotelio es normal.
Microscopía confocal
Observación de capas profundas: Se observan micropartículas altamente reflectantes dentro de los queratocitos anteriores a la membrana de Descemet.
Diagnóstico diferencial: Útil para distinguir de distrofias y degeneraciones corneales similares.
Otros diagnósticos diferenciales incluyen la distrofia corneal macular, la distrofia filiforme profunda y la distrofia puntiforme posterior. Todas presentan opacidades en la capa profunda de la córnea, pero la morfología y distribución de las opacidades difieren.
La diferenciación de la distrofia endotelial de Fuchs puede ser difícil solo con microscopía de lámpara de hendidura, y es necesaria la microscopía especular. En la degeneración corneal farinata, no se observan anomalías en el endotelio corneal.
Q¿Cuál es la diferencia con la distrofia endotelial de Fuchs?
A
Tanto la degeneración corneal farinata como la distrofia endotelial de Fuchs presentan hallazgos en las capas profundas de la córnea, pero la diferencia decisiva es el estado del endotelio corneal. En la degeneración corneal farinata, la microscopía especular muestra morfología y densidad normales de las células endoteliales, mientras que en Fuchs se observan gutas en la membrana de Descemet y disminución de la densidad celular endotelial. Fuchs progresa a edema corneal y queratopatía bullosa, pero la degeneración corneal farinata no afecta la visión.
La degeneración corneal farinata no requiere tratamiento. Dado que no afecta la visión ni presenta síntomas, solo se necesita seguimiento observacional.
En casos asociados con ictiosis ligada al cromosoma X, tampoco es necesario tratamiento para los hallazgos corneales. El manejo dermatológico es el principal.
Al ser una enfermedad degenerativa y de base genética, actualmente no existen métodos de prevención primaria.
Con el envejecimiento, se acumulan inclusiones similares a lipofuscina en el citoplasma de los queratocitos del estroma corneal justo anteriores a la membrana de Descemet. Histopatológicamente, se observan como vacuolas intracitoplasmáticas que contienen inclusiones similares a lipofuscina y pueden causar un agrandamiento anormal de los queratocitos.
La lipofuscina es un producto de la peroxidación lipídica debido al estrés oxidativo intracelular, y su acumulación progresa con la edad. El mecanismo de acumulación selectiva en los queratocitos del estroma profundo no se comprende completamente.
La deficiencia de esteroide sulfatasa debida a mutaciones en el gen STS causa un trastorno en el metabolismo del sulfato de colesterol. El sulfato de colesterol acumulado se deposita en los queratocitos, presentando una opacidad pulverulenta similar a la degeneración corneal farinata relacionada con la edad. STS se localiza en el retículo endoplásmico y puede estar involucrado en la formación de los depósitos similares a lipofuscina observados histopatológicamente.
Los depósitos corneales en la XLI tienden a aparecer a una edad más temprana que los cambios relacionados con la edad y se distribuyen más ampliamente. Se ha sugerido que la XLI y la degeneración corneal fariniforme relacionada con la edad pueden compartir una base fisiopatológica común, pero se necesita más investigación para una elucidación detallada 3).
Kobayashi A, Ohkubo S, Tagawa S, Uchiyama K, Sugiyama K. In vivo confocal microscopy in the patients with cornea farinata. Cornea. 2003;22(6):578-581.
Costagliola C, Fabbrocini G, Illiano GM, Scibelli G, Delfino M. Ocular findings in X-linked ichthyosis: a survey on 38 cases. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde. 1991;202(3):152-5. doi:10.1159/000310197. PMID:1923309.
Hung C, Ayabe RI, Wang C, Frausto RF, Aldave AJ. Pre-Descemet corneal dystrophy and X-linked ichthyosis associated with deletion of Xp22.31 containing the STS gene. Cornea. 2013;32(9):1283-7. doi:10.1097/ICO.0b013e318298e176. PMID:23807007; PMCID:PMC3740086.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.