La degenerazione farinosa della cornea (cornea farinata) è un reperto caratterizzato dalla comparsa di fini opacità polverulente immediatamente anteriormente alla membrana di Descemet, nello strato più profondo dello stroma corneale1). Fu descritta per la prima volta dall’oftalmologo svizzero Arthur Vogt ed è anche chiamata “cornea farinosa”.
Insorge bilateralmente e progredisce lentamente con l’età. Poiché non influisce sulla vista, il suo significato clinico è limitato. Viene classificata come degenerazione e non come distrofia corneale. Le segnalazioni al di sotto dei 40 anni sono rare. Alla microscopia confocale in vivo (IVCM) si osservano microparticelle altamente riflettenti nel citoplasma dei cheratociti dello stroma profondo 1).
Opacità stromali profonde simili si riscontrano anche nei pazienti con ittiosi legata all’X (XLI) dovuta a mutazione del gene STS (Xp22.31) 2,3). Sono presenti nel 50% dei pazienti e nel 25% delle donne portatrici e compaiono già in età adulta precoce 2). In questo caso non sono legate all’invecchiamento ma a un deficit di steroido solfatasi, con conseguente accumulo di colesterolo solfato 3).
QLa degenerazione farinosa della cornea necessita di trattamento?
A
La degenerazione farinosa della cornea di solito non richiede trattamento. Essendo asintomatica e senza effetti sulla vista, è sufficiente la sola osservazione. Tuttavia, è importante differenziarla da altre condizioni con opacità stromali profonde simili, come la distrofia endoteliale di Fuchs. Nella distrofia di Fuchs si osserva una riduzione della densità delle cellule endoteliali, mentre nella degenerazione farinosa della cornea l’endotelio è normale.
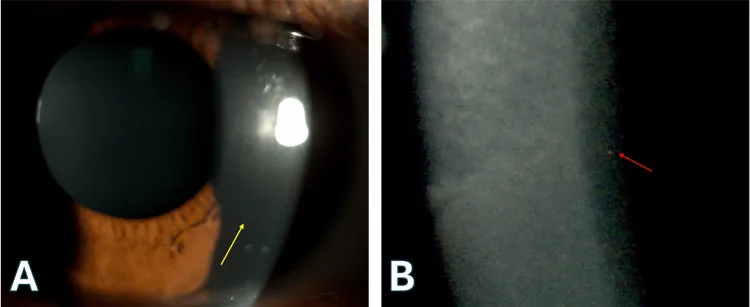
Joobin Khadamy Ocular Manifestations Leading to the Diagnosis of Ichthyosis: A Case Report 2025 Mar 4 Cureus.; 17(3):e80023 Figure 2. PMCID: PMC11968076. License: CC BY.
L’immagine A è una fotografia dell’occhio acquisita con microscopia a lampada a fessura, che mostra nervi corneali sollevati (frecce gialle). L’immagine B è una vista ingrandita con microscopia a lampada a fessura, che mostra opacità punteggiate nello stroma corneale profondo (frecce rosse). Questi reperti sono compatibili con la diagnosi di cornea farinata.
La degenerazione farinosa della cornea è solitamente asintomatica. Non causa riduzione della vista, dolore oculare, sensazione di corpo estraneo o fotofobia. Nella maggior parte dei casi viene scoperta incidentalmente durante l’esame con lampada a fessura.
Alla lampada a fessura, in retroilluminazione o a riflessione speculare, si osservano fini depositi granulari grigio-biancastri o giallo-brunastri sulla superficie posteriore della cornea. Le opacità sono dense nelle aree centrale e paracentrale e diminuiscono verso la periferia. Ogni deposito è estremamente piccolo e può essere facilmente trascurato con l’illuminazione diretta.
Le opacità sono distribuite in modo diffuso e uniforme nello stroma corneale immediatamente anteriormente alla membrana di Descemet. La membrana di Descemet, l’epitelio corneale e lo strato di cellule endoteliali sono normali. Lo spessore corneale è nei limiti normali. La microscopia speculare mostra morfologia e densità normali delle cellule endoteliali.
Il principale fattore di rischio è l’età. È comune negli anziani e progredisce lentamente, ma raramente è clinicamente problematico. I casi sotto i 40 anni sono rari.
Il deficit di steroidi solfatasi dovuto a una mutazione del gene STS (Xp22.31) causa l’ittiosi legata all’X (XLI). Nei pazienti con XLI, il colesterolo solfato si accumula nello stroma corneale profondo, provocando un’opacità simile alla degenerazione farinosa della cornea. Sono state riportate almeno sei diverse mutazioni del gene STS e il tipo di mutazione influenza l’espressione e la funzione dell’enzima steroide solfatasi, portando a fenotipi vari.
Retroilluminazione : metodo di base per visualizzare i fini depositi granulari sulla superficie posteriore della cornea.
Riflesso speculare : consente di osservare l’opacità appena prima della membrana di Descemet con alto contrasto.
Illuminazione diretta : i piccoli depositi sono facili da trascurare, quindi è importante combinarla con la retroilluminazione.
Microscopia speculare
Valutazione endoteliale : confermare che la morfologia e la densità delle cellule endoteliali siano normali.
Diagnosi differenziale con Fuchs : nella distrofia di Fuchs si osservano guttae e riduzione della densità delle cellule endoteliali, mentre in questa malattia l’endotelio è normale.
Microscopia confocale
Osservazione profonda : si osservano microparticelle altamente riflettenti nei cheratociti dello stroma corneale anteriormente alla membrana di Descemet.
Diagnosi differenziale: utile per distinguere distrofie corneali simili e degenerazioni.
Altre diagnosi differenziali includono la distrofia corneale a macchie (fleck corneal dystrophy), la distrofia filiforme profonda (deep filiform dystrophy) e la distrofia puntiforme posteriore (posterior punctiform dystrophy). Tutte presentano opacità negli strati profondi della cornea, ma la morfologia e la distribuzione delle opacità differiscono.
La distinzione tra la distrofia endoteliale di Fuchs e la degenerazione farinosa della cornea può essere difficile con il solo microscopio a lampada a fessura; è necessaria la microscopia speculare. Nella degenerazione farinosa della cornea, l’endotelio corneale è normale.
QQual è la differenza rispetto alla distrofia endoteliale di Fuchs?
A
Sia la degenerazione farinosa della cornea che la distrofia endoteliale di Fuchs presentano reperti negli strati profondi della cornea, ma la differenza cruciale è lo stato dell’endotelio corneale. Nella degenerazione farinosa della cornea, la microscopia speculare mostra una morfologia e densità normali delle cellule endoteliali, mentre nella distrofia di Fuchs si osservano guttae (escrescenze verrucose) sulla membrana di Descemet e una ridotta densità delle cellule endoteliali. La distrofia di Fuchs può progredire fino all’edema corneale e alla cheratopatia bollosa, mentre la degenerazione farinosa della cornea non influisce sulla vista.
La degenerazione farinosa della cornea non richiede trattamento. Poiché non influisce sulla vista e non causa sintomi, è sufficiente un semplice follow-up.
Anche in caso di associazione con ittiosi legata all’X, non è necessario alcun trattamento per i reperti corneali. La gestione dermatologica è prioritaria.
Poiché si tratta di una malattia degenerativa e su base genetica, attualmente non esiste un metodo di prevenzione primaria.
Con l’avanzare dell’età, inclusioni simili alla lipofuscina si accumulano nel citoplasma dei cheratociti dello stroma corneale immediatamente prima della membrana di Descemet. Istopatologicamente, si osservano come vacuoli citoplasmatici contenenti inclusioni simili alla lipofuscina, che possono causare un’ipertrofia anomala dei cheratociti.
La lipofuscina è un prodotto della perossidazione lipidica dovuta allo stress ossidativo cellulare, il cui accumulo aumenta con l’età. Il meccanismo di accumulo selettivo nei cheratociti dello stroma profondo non è completamente chiarito.
Il deficit di steroidi sulfatasi dovuto a una mutazione del gene STS causa un’alterazione del metabolismo del colesterolo solfato. Il colesterolo solfato accumulato si deposita nei cheratociti dello stroma corneale, provocando un’opacità polverosa simile alla degenerazione farinosa della cornea legata all’età. La STS è localizzata nel reticolo endoplasmatico delle cellule e potrebbe essere coinvolta nella formazione dei depositi simili alla lipofuscina osservati in istopatologia.
Nella XLI, i depositi corneali compaiono più precocemente rispetto ai cambiamenti legati all’età e tendono ad essere più estesi. È stato suggerito che la XLI e la degenerazione corneale farinosa legata all’età possano condividere una base fisiopatologica comune, ma sono necessarie ulteriori ricerche per un chiarimento dettagliato 3).
Kobayashi A, Ohkubo S, Tagawa S, Uchiyama K, Sugiyama K. In vivo confocal microscopy in the patients with cornea farinata. Cornea. 2003;22(6):578-581.
Costagliola C, Fabbrocini G, Illiano GM, Scibelli G, Delfino M. Ocular findings in X-linked ichthyosis: a survey on 38 cases. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde. 1991;202(3):152-5. doi:10.1159/000310197. PMID:1923309.
Hung C, Ayabe RI, Wang C, Frausto RF, Aldave AJ. Pre-Descemet corneal dystrophy and X-linked ichthyosis associated with deletion of Xp22.31 containing the STS gene. Cornea. 2013;32(9):1283-7. doi:10.1097/ICO.0b013e318298e176. PMID:23807007; PMCID:PMC3740086.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.